 +1 credit
+1 credit
Video Upload Options
Asymmetric cell division (ACD) of neural stem cells and progenitors not only renews the stem cell population but also ensures the normal development of the nervous system, producing various types of neurons with different shapes and functions in the brain. One major mechanism to achieve ACD is the asymmetric localization and uneven segregation of intracellular proteins and organelles into sibling cells. Recent studies have demonstrated that liquid-liquid phase separation (LLPS) provides a potential mechanism for the formation of membrane-less biomolecular condensates that are asymmetrically distributed on limited membrane regions. Moreover, mechanical forces have emerged as pivotal regulators of asymmetric neural stem cell division by generating sibling cell size asymmetry. In this review, we will summarize recent discoveries of ACD mechanisms driven by LLPS and mechanical forces.
1. Asymmetric Cell Division of Drosophila Neuroblasts
1.1. Asymmetric Protein Localization during ACD
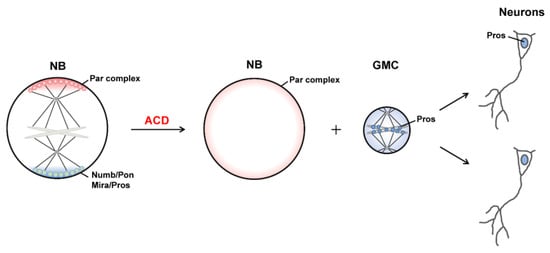
1.2. Generation of Distinct Sibling Cells
2. LLPS and Asymmetric Protein Localization during ACD of NBs
2.1. LLPS in Cells
2.2. LLPS-Mediated Basal Localization of Numb and Pon in Dividing NBs
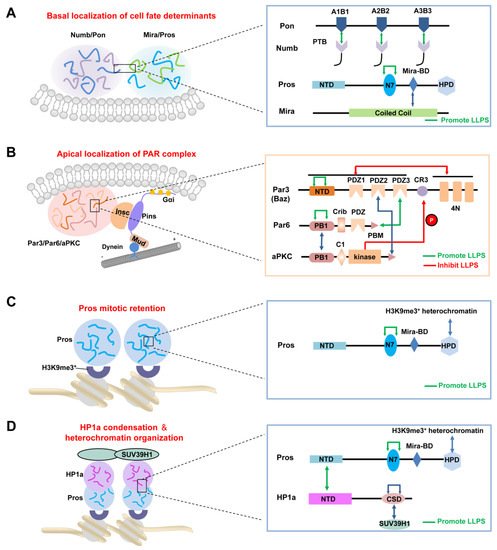
2.3. LLPS-Mediated Apical Localization of the PAR Complexes in Dividing NBs
2.4. LLPS-mediated mitotic implantation of Pros in dividing GMCs
After cytokinesis, the transcription factor Pros enters the GMC nucleus to promote its differentiation [57,58]. Recently, Liu et al. showed that Pros drives irreversible terminal neuronal differentiation by regulating heterochromatin domain condensation and expansion in an LLPS-dependent manner (Figure 2C) [59]. Pros was found to undergo LLPS in vitro and in vivo through self-association through its N7 motif (Figure 2C). LLPS of Pros enabled its retention at histone H3 Lys9 tri-methylation (H3K9me3) heterochromatin regions of chromosomes in mitotic GMCs, where it recruited and concentrated the H3K9me3 “reader” heterochromatin protein 1 (HP1) into the condensed phase via its N-terminal domain (Figure 2D), thus driving the condensation and expansion of the H3K9me3+ heterochromatin regions in the newly generated neurons. After HP1 condensation, Pros, together with a portion of HP1, detached from the H3K9me3+ heterochromatin regions and translocated to its target gene loci, where Pros and HP1 acted cooperatively to silence Pros target genes permanently to drive cell-cycle exit and terminal neuronal differentiation [59]. Pros mutants that exhibited impaired LLPS ability prevented Pros from being retained on chromosomes and thus resulted in compromised terminal differentiation. The above phenotype could be effectively rescued by replacing the N7 motif with another IDR protein capable of LLPS. Interestingly, though the recombinant N7-containing Pros fragment and HP1a co-phase separate in vitro, the Pros condensates and HP1a condensates do not coalesce in vivo [59], suggesting the existence of unknown regulating mechanism(s). Moreover, it is plausible that the basal distribution of Pros in dividing NBs might also be driven by its phase separation, together with the Mira dimer via its coiled-coil domain (Figure 2A).
3. Mechanical Forces Regulating ACD
3.1. Polarity Cue-Regulated Spindle Orientation
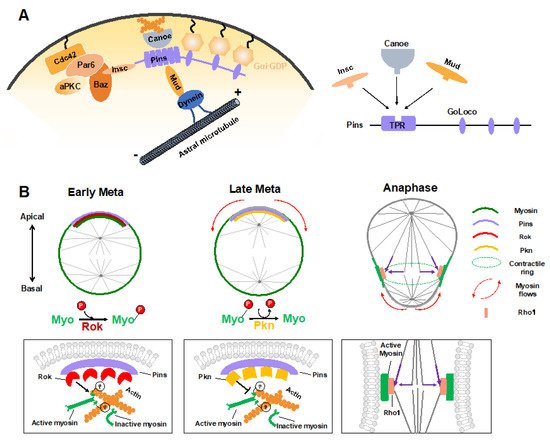
3.2. Myosin Flows Regulated by Polarity and Spindle Cues
References
- Delgado, M.K.; Cabernard, C. Mechanical regulation of cell size, fate, and behavior during asymmetric cell division. Curr. Opin. Cell Biol. 2020, 67, 9–16.
- Speicher, S.; Fischer, A.; Knoblich, J.; Carmena, A. The PDZ Protein Canoe Regulates the Asymmetric Division of Drosophila Neuroblasts and Muscle Progenitors. Curr. Biol. 2008, 18, 831–837.
- Wee, B.; Johnston, C.A.; Prehoda, K.E.; Doe, C.Q. Canoe binds RanGTP to promote PinsTPR/Mud-mediated spindle orientation. J. Cell Biol. 2011, 195, 369–376.
- Choi, W.; Acharya, B.; Peyret, G.; Fardin, M.-A.; Mège, R.-M.; Ladoux, B.; Yap, A.; Fanning, A.S.; Peifer, M. Remodeling the zonula adherens in response to tension and the role of afadin in this response. J. Cell Biol. 2016, 213, 243–260.
- Gao, L.; Yang, Z.; Hiremath, C.; Zimmerman, S.E.; Long, B.; Brakeman, P.R.; Mostov, K.E.; Bryant, D.M.; Luby-Phelps, K.; Marciano, D.K. Developing renal tubules orient cell division via Afadin to position the tubule lumen. Development 2017, 144, 3511–3520.
- Keder, A.; Rives-Quinto, N.; Aerne, B.L.; Franco, M.; Tapon, N.; Carmena, A. The Hippo Pathway Core Cassette Regulates Asymmetric Cell Division. Curr. Biol. 2015, 25, 2739–2750.
- Dewey, E.; Sanchez, D.; Johnston, C.A. Warts Phosphorylates Mud to Promote Pins-Mediated Mitotic Spindle Orientation in Drosophila, Independent of Yorkie. Curr. Biol. 2015, 25, 2751–2762.
- Zhu, J.; Wen, W.; Zheng, Z.; Shang, Y.; Wei, Z.; Xiao, Z.; Pan, Z.; Du, Q.; Wang, W.; Zhang, M. LGN/mInsc and LGN/NuMA Complex Structures Suggest Distinct Functions in Asymmetric Cell Division for the Par3/mInsc/LGN and Gαi/LGN/NuMA Pathways. Mol. Cell 2011, 43, 418–431.
- Carminati, M.; Gallini, S.; Pirovano, L.; Alfieri, A.; Bisi, S.; Mapelli, M. Concomitant binding of Afadin to LGN and F-actin directs planar spindle orientation. Nat. Struct. Mol. Biol. 2016, 23, 155–163.
- Gloerich, M.; Bianchini, J.M.; Siemers, K.A.; Cohen, D.J.; Nelson, W.J. Cell division orientation is coupled to cell–cell adhesion by the E-cadherin/LGN complex. Nat. Commun. 2017, 8, 13996.
- Finegan, T.M.; Bergstralh, D.T. Division orientation: Disentangling shape and mechanical forces. Cell Cycle 2019, 18, 1187–1198.
- Connell, M.; Cabernard, C.; Ricketson, D.; Doe, C.Q.; Prehoda, K.E. Asymmetric cortical extension shifts cleavage furrow position in Drosophila neuroblasts. Mol. Biol. Cell 2011, 22, 4220–4226.
- Cabernard, C.; Prehoda, K.E.; Doe, C.Q. A spindle-independent cleavage furrow positioning pathway. Nat. Cell Biol. 2010, 467, 91–94.
- Tsankova, A.; Pham, T.; Garcia, D.S.; Otte, F.; Cabernard, C. Cell Polarity Regulates Biased Myosin Activity and Dynamics during Asymmetric Cell Division via Drosophila Rho Kinase and Protein Kinase N. Dev. Cell 2017, 42, 143–155.e5.
- Ou, G.; Stuurman, N.; D’Ambrosio, M.; Vale, R.D. Polarized Myosin Produces Unequal-Size Daughters during Asymmetric Cell Division. Science 2010, 330, 677–680.
- Roubinet, C.; Tsankova, A.; Pham, T.; Monnard, A.; Caussinus, E.; Affolter, M.; Cabernard, C. Spatio-temporally separated cortical flows and spindle geometry establish physical asymmetry in fly neural stem cells. Nat. Commun. 2017, 8, 1–16.
- Mayer, M.; Depken, M.; Bois, J.; Julicher, F.; Grill, S.W. Anisotropies in cortical tension reveal the physical basis of polarizing cortical flows. Nat. Cell Biol. 2010, 467, 617–621.
- Pham, T.T.; Monnard, A.; Helenius, J.; Lund, E.; Lee, N.; Müller, D.J.; Cabernard, C. Spatiotemporally Controlled Myosin Relocalization and Internal Pressure Generate Sibling Cell Size Asymmetry. iScience 2019, 13, 9–19.
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; van den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435.
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213.
- Roden, C.; Gladfelter, A.S. RNA contributions to the form and function of biomolecular condensates. Nat. Rev. Mol. Cell Biol. 2021, 22, 183–195.
- Hyman, A.A.; Weber, C.A.; Julicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58.
- Zhao, Y.G.; Zhang, H. Phase Separation in Membrane Biology: The Interplay between Membrane-Bound Organelles and Membraneless Condensates. Dev. Cell 2020, 55, 30–44.
- Quiroz, F.G.; Fiore, V.F.; Levorse, J.; Polak, L.; Wong, E.; Pasolli, H.A.; Fuchs, E. Liquid-liquid phase separation drives skin barrier formation. Science 2020, 367, eaax9554.
- Ong, J.Y.; Torres, J.Z. Phase Separation in Cell Division. Mol. Cell 2020, 80, 9–20.
- Wu, X.; Cai, Q.; Feng, Z.; Zhang, M. Liquid-Liquid Phase Separation in Neuronal Development and Synaptic Signaling. Dev. Cell 2020, 55, 18–29.
- Tsang, B.; Pritišanac, I.; Scherer, S.W.; Moses, A.M.; Forman-Kay, J.D. Phase Separation as a Missing Mechanism for Interpretation of Disease Mutations. Cell 2020, 183, 1742–1756.
- Zhang, H.; Ji, X.; Li, P.; Liu, C.; Lou, J.; Wang, Z.; Wen, W.; Xiao, Y.; Zhang, M.; Zhu, X. Liquid-liquid phase separation in biology: Mechanisms, physiological functions and human diseases. Sci. China Life Sci. 2020, 63, 953–985.
- Jain, A.; Vale, A.J.R.D. RNA phase transitions in repeat expansion disorders. Nat. Cell Biol. 2017, 546, 243–247.
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077.
- Wang, Z.; Zhang, H. Phase Separation, Transition, and Autophagic Degradation of Proteins in Development and Pathogenesis. Trends Cell Biol. 2019, 29, 417–427.
- Wright, P.E.; Dyson, H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29.
- Posey, A.E.; Holehouse, A.S.; Pappu, R.V. Phase Separation of Intrinsically Disordered Proteins. Methods Enzymol. 2018, 611, 1–30.
- Banani, S.F.; Lee, H.O.; Hyman, A.A.; Rosen, M.K. Biomolecular condensates: Organizers of cellular biochemistry. Nat. Rev. Mol. Cell Biol. 2017, 18, 285–298.
- Banani, S.F.; Rice, A.; Peeples, W.B.; Lin, Y.; Jain, S.; Parker, R.; Rosen, M.K. Compositional Control of Phase-Separated Cellular Bodies. Cell 2016, 166, 651–663.
- Zeng, M.; Shang, Y.; Araki, Y.; Guo, T.; Huganir, R.L.; Zhang, M. Phase Transition in Postsynaptic Densities Underlies Formation of Synaptic Complexes and Synaptic Plasticity. Cell 2016, 166, 1163–1175.e12.
- Li, P.; Banjade, S.; Cheng, H.-C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nat. Cell Biol. 2012, 483, 336–340.
- Nott, T.; Petsalaki, E.; Farber, P.; Jervis, D.; Fussner, E.; Plochowietz, A.; Craggs, T.; Bazett-Jones, D.P.; Pawson, T.; Forman-Kay, J.D.; et al. Phase Transition of a Disordered Nuage Protein Generates Environmentally Responsive Membraneless Organelles. Mol. Cell 2015, 57, 936–947.
- Shan, Z.; Tu, Y.; Yang, Y.; Liu, Z.; Zeng, M.; Xu, H.; Long, J.; Zhang, M.; Cai, Y.; Wen, W. Basal condensation of Numb and Pon complex via phase transition during Drosophila neuroblast asymmetric division. Nat. Commun. 2018, 9, 1–16.
- Liu, Z.; Yang, Y.; Gu, A.; Xu, J.; Mao, Y.; Lu, H.; Hu, W.; Lei, Q.-Y.; Li, Z.; Zhang, M.; et al. Par complex cluster formation mediated by phase separation. Nat. Commun. 2020, 11, 1–18.
- Lu, B.; Ackerman, L.; Jan, L.; Jan, Y.-N. Modes of Protein Movement that Lead to the Asymmetric Localization of Partner of Numb during Drosophila Neuroblast Division. Mol. Cell 1999, 4, 883–891.
- Patel, S.S.; Belmont, B.; Sante, J.M.; Rexach, M.F. Natively Unfolded Nucleoporins Gate Protein Diffusion across the Nuclear Pore Complex. Cell 2007, 129, 83–96.
- Kono, K.; Yoshiura, S.; Fujita, I.; Okada, Y.; Shitamukai, A.; Shibata, T.; Matsuzaki, F. Reconstruction of Par-dependent polarity in apolar cells reveals a dynamic process of cortical polarization. eLife 2019, 8, 8.
- Wilson, M.I.; Gill, D.J.; Perisic, O.; Quinn, M.; Williams, R.L. PB1 Domain-Mediated Heterodimerization in NADPH Oxidase and Signaling Complexes of Atypical Protein Kinase C with Par6 and p62. Mol. Cell 2003, 12, 39–50.
- Soriano, E.V.; Ivanova, M.E.; Fletcher, G.; Riou, P.; Knowles, P.P.; Barnouin, K.; Purkiss, A.; Kostelecky, B.; Saiu, P.; Linch, M.; et al. aPKC Inhibition by Par3 CR3 Flanking Regions Controls Substrate Access and Underpins Apical-Junctional Polarization. Dev. Cell 2016, 38, 384–398.
- Holly, R.W.; Jones, K.; Prehoda, K.E. A Conserved PDZ-Binding Motif in aPKC Interacts with Par-3 and Mediates Cortical Polarity. Curr. Biol. 2020, 30, 893–898.e5.
- De Sá, E.M.; Mirouse, V.; Johnston, D.S. aPKC Phosphorylation of Bazooka Defines the Apical/Lateral Border in Drosophila Epithelial Cells. Cell 2010, 141, 509–523.
- Wang, S.-C.; Low, T.Y.F.; Nishimura, Y.; Gole, L.; Yukako, N.; Motegi, F. Cortical forces and CDC-42 control clustering of PAR proteins for Caenorhabditis elegans embryonic polarization. Nat. Cell Biol. 2017, 19, 988–995.
- Rodriguez, J.; Peglion, F.; Martin, J.; Hubatsch, L.; Reich, J.; Hirani, N.; Gubieda, A.G.; Roffey, J.; Fernandes, A.R.; Johnston, D.S.; et al. aPKC Cycles between Functionally Distinct PAR Protein Assemblies to Drive Cell Polarity. Dev. Cell 2017, 42, 400–415.e9.
- Oon, C.H.; Prehoda, K.E. Asymmetric recruitment and actin-dependent cortical flows drive the neuroblast polarity cycle. eLife 2019, 8, 8.

