 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ryszard Pluta | + 1990 word(s) | 1990 | 2021-09-01 08:57:54 | | | |
| 2 | Jessie Wu | Meta information modification | 1990 | 2021-10-29 04:13:08 | | |
Video Upload Options
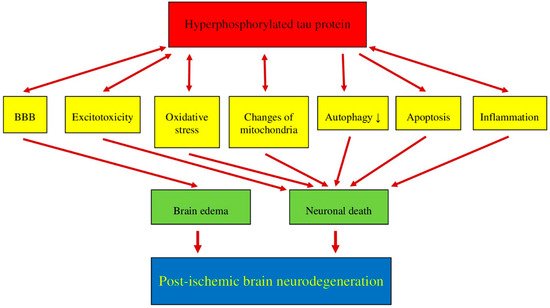
Recent data suggest that post-ischemic brain neurodegeneration in humans and animals is associated with the modified tau protein in a manner typical of Alzheimer’s disease neuropathology. Pathological changes in the tau protein, at the gene and protein level due to cerebral ischemia, can lead to the development of Alzheimer’s disease-type neuropathology and dementia. Some studies have shown increased tau protein staining and gene expression in neurons following ischemia-reperfusion brain injury. Recent studies have found the tau protein to be associated with oxidative stress, apoptosis, autophagy, excitotoxicity, neuroinflammation, blood-brain barrier permeability, mitochondrial dysfunction, and impaired neuronal function. In this review, we discuss the interrelationship of these phenomena with post-ischemic changes in the tau protein in the brain. The tau protein may be at the intersection of many pathological mechanisms due to severe neuropathological changes in the brain following ischemia. The data indicate that an episode of cerebral ischemia activates the damage and death of neurons in the hippocampus in a tau protein-dependent manner, thus determining a novel and important mechanism for the survival and/or death of neuronal cells following ischemia. In this review, we update our understanding of proteomic and genomic changes in the tau protein in post-ischemic brain injury and present the relationship between the modified tau protein and post-ischemic neuropathology and present a positive correlation between the modified tau protein and a post-ischemic neuropathology that has characteristics of Alzheimer’s disease-type neurodegeneration.
1. Post-Ischemic Tau Protein versus Blood-Brain Barrier
2. Post-Ischemic Tau Protein versus Excitotoxicity
3. Post-Ischemic Tau Protein versus Oxidative Stress
4. Post-Ischemic Tau Protein versus Mitochondria
5. Post-Ischemic Tau Protein versus Autophagy
6. Post-Ischemic Tau Protein versus Apoptosis
7. Post-Ischemic Tau Protein versus Neuroinflammation
References
- Majd, S.; Power, J.H.; Koblar, S.A.; Grantham, H.J.M. Introducing a developed model of reversible cardiac arrest to produce global brain ischemia and its impact on microtubule-associated protein tau phosphorylation at Ser396. Int. J. Neurol. Neurother. 2016, 3, 040.
- Fujii, H.; Takahashi, T.; Mukai, T.; Tanaka, S.; Hosomi, N.; Maruyama, H.; Sakai, N.; Matsumoto, M. Modifications of tau protein after cerebral ischemia and reperfusion in rats are similar to those occurring in Alzheimer’s disease—Hyperphosphorylation and cleavage of 4- and 3-repeat tau. Br. J. Pharmacol. 2016, 37, 2441–2457.
- Wen, Y.; Yang, S.; Liu, R.; Simpkins, J.W. Transient cerebral ischemia induces site-specific hyperphosphorylation of tau protein. Brain Res. 2004, 1022, 30–38.
- Wen, Y.; Yang, S.; Liu, R.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Transient Cerebral Ischemia Induces Aberrant Neuronal Cell Cycle Re-entry and Alzheimer’s Disease-like Tauopathy in Female Rats. J. Biol. Chem. 2004, 279, 22684–22692.
- Wen, Y.; Yang, S.-H.; Liu, R.; Perez, E.J.; Brun-Zinkernagel, A.M.; Koulen, P.; Simpkins, J.W. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2007, 1772, 473–483.
- Majd, S.; Power, J.H.T.; Koblar, S.; Grantham, H. Early glycogen synthase kinase-3β and protein phosphatase 2A independent tau dephosphorylation during global brain ischaemia and reperfusion following cardiac arrest and the role of the adenosine monophosphate kinase pathway. Eur. J. Neurosci. 2016, 44, 1987–1997.
- Kovalska, M.; Tothova, B.; Kovalska, L.; Tatarkova, Z.; Kalenska, D.; Tomascova, A.; Adamkov, M.; Lehotsky, J. Association of Induced Hyperhomocysteinemia with Alzheimer’s Disease-Like Neurodegeneration in Rat Cortical Neurons After Global Ischemia-Reperfusion Injury. Neurochem. Res. 2018, 43, 1766–1778.
- Kato, T.; Hirano, A.; Katagiri, T.; Sasaki, H.; Yamada, S. Neurofibrillary tangle formation in the nucleus basalis of meynert ipsilateral to a massive cerebral infarct. Ann. Neurol. 1988, 23, 620–623.
- Hatsuta, H.; Takao, M.; Nogami, A.; Uchino, A.; Sumikura, H.; Takata, T.; Morimoto, S.; Kanemaru, K.; Adachi, T.; Arai, T.; et al. Tau and TDP-43 accumulation of the basal nucleus of Meynert in individuals with cerebral lobar infarcts or hemorrhage. Acta Neuropathol. Commun. 2019, 7, 49.
- Tuo, Q.Z.; Lei, P.; Jackman, K.A.; Li, X.L.; Xiong, H.; Li, X.L.; Liuyang, Z.Y.; Roisman, L.; Zhang, S.T.; Ayton, S.; et al. Tau mediated iron export prevents ferroptotic damage after ischemic stroke. Mol. Psychiatry 2017, 22, 1520–1530.
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Muller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473.
- Pluta, R.; Lossinsky, A.; Wisniewski, H.; Mossakowski, M. Early blood-brain barrier changes in the rat following transient complete cerebral ischemia induced by cardiac arrest. Brain Res. 1994, 633, 41–52.
- Pluta, R. Blood-brain barrier dysfunction and amyloid precursor protein accumulation in microvascular compartment following ischemia-reperfusion brain injury with 1-year survival. Acta Neurochir. Suppl. 2003, 86, 117–122.
- Pluta, R. Pathological Opening of the Blood-Brain Barrier to Horseradish Peroxidase and Amyloid Precursor Protein following Ischemia-Reperfusion Brain Injury. Chemotherapy 2005, 51, 223–226.
- Pluta, R.; Ułamek, M.; Januszewski, S. Micro-blood-brain barrier openings and cytotoxic fragments of amyloid precursor protein accumulation in white matter after ischemic brain injury in long-lived rats. Pain 2006, 96, 267–271.
- Pluta, R.; Januszewski, S.; Ulamek, M. Ischemic blood-brain barrier and amyloid in white matter as etiological factors in leukoaraiosis. Acta Neurochir. Suppl. 2008, 102, 353–356.
- Basurto-Islas, G.; Gu, J.-H.; Tung, Y.C.; Liu, F.; Iqbal, K. Mechanism of Tau Hyperphosphorylation Involving Lysosomal Enzyme Asparagine Endopeptidase in a Mouse Model of Brain Ischemia. J. Alzheimers Dis. 2018, 63, 821–833.
- Khan, S.; Yuldasheva, N.Y.; Batten, T.F.C.; Pickles, A.R.; Kellett, K.A.B.; Saha, S. Tau pathology and neurochemical changes associ-ated with memory dysfunction in an optimized murine model of global cerebral ischaemia—A potential model for vascular dementia? Neurochem. Int. 2018, 118, 134–144.
- Ramos-Cejudo, J.; Wisniewski, T.; Marmar, C.; Zetterberg, H.; Blennow, K.; de Leon, M.J.; Fossati, S. Traumatic brain injury and Alzheimer’s disease: The cerebrovascular link. EBio Med. 2018, 28, 21–30.
- Lee, P.H.; Bang, O.Y.; Hwang, E.M.; Lee, J.S.; Joo, U.S.; Mook-Jung, I.; Huh, K. Circulating beta amyloid protein is elevated in patients with acute ischemic stroke. J. Neural. Transm. 2005, 112, 1371–1379.
- Zetterberg, H.; Mortberg, E.; Song, L.; Chang, L.; Provuncher, G.K.; Patel, P.P.; Ferrell, E.; Fournier, D.R.; Kan, C.W.; Campbell, T.G.; et al. Hypoxia due to cardiac arrest induces a time-dependent increase in serum amyloidβ levels in humans. PLoS ONE 2011, 6, e28263.
- Li, P.; Stetler, R.A.; Leak, R.; Shi, Y.; Li, Y.; Yu, W.; Bennett, M.V.; Chen, J. Oxidative stress and DNA damage after cerebral ischemia: Potential therapeutic targets to repair the genome and improve stroke recovery. Neuropharmacology 2017, 134, 208–217.
- Sekeljic, V.; Bataveljic, D.; Stamenkovic, S.; Ułamek, M.; Jabłoński, M.; Radenovic, L.; Pluta, R.; Andjus, P.R. Cellular markers of neu-roinflammation and neurogenesis after ischemic brain injury in the long-term survival rat model. Brain Struct. Funct. 2012, 217, 411–420.
- Radenovic, L.; Nenadic, M.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J.; Andjus, P.R.; Pluta, R. Heterogeneity in brain distribution of activated microglia and astrocytes in a rat ischemic model of Alzheimer’s disease after 2 years of survival. Aging 2020, 12, 12251–12267.
- Kumfu, S.; Charununtakorn, S.T.; Jaiwongkam, T.; Chattipakorn, N.; Chattipakorn, S.C. Humanin Exerts Neuroprotection During Cardiac Ischemia-Reperfusion Injury. J. Alzheimer’s Dis. 2018, 61, 1343–1353.
- Mörtberg, E.; Zetterberg, H.; Nordmark, J.; Blennow, K.; Catry, C.; Decraemer, H.; Vanmechelen, E.; Rubertsson, S. Plasma tau protein in comatose patients after cardiac arrest treated with therapeutic hypothermia. Acta Anaesthesiol. Scand. 2011, 55, 1132–1138.
- Randall, J.; Mörtberg, E.; Provuncher, G.K.; Fournier, D.R.; Duffy, D.C.; Rubertsson, S.; Blennow, K.; Zetterberg, H.; Wilson, D.H. Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: Results of a pilot study. Resuscitation 2012, 84, 351–356.
- Banks, W.A.; Kovac, A.; Majerova, P.; Bullock, K.M.; Shi, M.; Zhang, J. Tau Proteins Cross the Blood-Brain Barrier. J. Alzheimer’s Dis. 2016, 55, 411–419.
- Ueno, M.; Chiba, Y.; Murakami, R.; Matsumoto, K.; Kawauchi, M.; Fujihara, R. Blood-brain barrier and blood–cerebrospinal fluid barrier in normal and pathological conditions. Brain Tumor Pathol. 2016, 33, 89–96.
- Pluta, R.; Salínska, E.; Puka, M.; Stafiej, A.; Lazarewicz, J. Early changes in extracellular amino acids and calcium concentrations in rabbit hippocampus following complete 15-min cerebral ischemia. Resuscitation 1988, 16, 193–210.
- Ojo, O.B.; Amoo, Z.A.; Saliu, I.O.; Olaleye, M.T.; Farombi, E.O.; Akinmoladun, A.C. Neurotherapeutic potential of kolaviron on neurotransmitter dysregulation, excitotoxicity, mitochondrial electron transport chain dysfunction and redox imbalance in 2-VO brain ischemia/reperfusion injury. Biomed. Pharmacother. 2019, 111, 859–872.
- Tejeda, G.S.; Esteban-Ortega, G.M.; San Antonio, E.; Vidaurre, O.G.; Díaz-Guerra, M. Prevention of excitotoxicity-induced processing of BDNF receptor TrkB-FL leads to stroke neuroprotection. EMBO Mol. Med. 2019, 11, e9950.
- Ho, P.I.; Ortiz, D.; Rogers, E.; Shea, T.B. Multiple aspects of homocysteine neurotoxicity: Glutamate excitotoxicity, kinase hyperactivation and DNA damage. J. Neurosci. Res. 2002, 70, 694–702.
- Ekinci, F.J.; Malik, K.U.; Shea, T.B. Activation of the L voltage-sensitive calcium channel by mitogen-activated protein (MAP) kinase following exposure of neuronal cells to beta-amyloid. MAP kinase mediates beta-amyloid-induced neurodegeneration. J. Biol. Chem. 1999, 274, 30322–30327.
- Petroni, D.; Tsai, J.; Mondal, D.; George, W. Attenuation of low dose methylmercury and glutamate induced-cytotoxicity and tau phosphorylation by anN-methyl-D-aspartate antagonist in human neuroblastoma (SHSY5Y) cells. Environ. Toxicol. 2011, 28, 700–706.
- Chen, X.; Jiang, H. Tau as a potential therapeutic target for ischemic stroke. Aging 2019, 11, 12827–12843.
- De Vos, A.; Bjerke, M.; Brouns, R.; De Roeck, N.; Jacobs, D.; Van den Abbeele, L.; Guldolf, K.; Zetterberg, H.; Blennow, K.; Engelborghs, S.; et al. Neurogranin and tau in cerebrospinal fluid and plasma of patients with acute ischemic stroke. BMC Neurol. 2017, 17, 170.
- Amadoro, G.; Ciotti, M.T.; Costanzi, M.; Cestari, V.; Calissano, P.; Canu, N. NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation. Proc. Natl. Acad. Sci. USA 2006, 103, 2892–2897.
- Hardingham, G.E.; Bading, H. Synaptic versus extrasynaptic NMDA receptor signalling: Implications for neurodegenerative disorders. Nat. Rev. Neurosci. 2010, 11, 682–696.
- Holth, J.K.; Bomben, V.C.; Reed, J.G.; Inoue, T.; Younkin, L.; Younkin, S.G.; Pautler, R.G.; Botas, J.; Noebels, J.L. Tau Loss Attenuates Neuronal Network Hyperexcitability in Mouse and Drosophila Genetic Models of Epilepsy. J. Neurosci. 2013, 33, 1651–1659.
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2012, 698, 6–18.
- Hunsberger, H.C.; Rudy, C.C.; Batten, S.R.; Gerhardt, G.A.; Reed, M.N. P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J. Neurochem. 2015, 132, 169–182.
- Pallo, S.P.; DiMaio, J.; Cook, A.; Nilsson, B.; Johnson, G.V. Mechanisms of tau and Aβ-induced excitotoxicity. Brain Res. 2015, 1634, 119–131.
- Decker, J.M.; Krüger, L.; Sydow, A.; Dennissen, F.J.; Siskova, Z.; Mandelkow, E.; Mandelkow, E.M. The Tau/A152T mutation, a risk factor for frontotemporal-spectrum disorders, leads to NR2B receptor-mediated excitotoxicity. EMBO Rep. 2016, 17, 552–569.
- Miyamoto, T.; Stein, L.; Thomas, R.; Djukic, B.; Taneja, P.; Knox, J.; Vossel, K.; Mucke, L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 2017, 12, 1–19.
- Zhou, S.; Yu, G.; Chi, L.; Zhu, J.; Zhang, W.; Zhang, Y.; Zhang, L. Neuroprotective effects of edaravone on cognitive deficit, oxidative stress and tau hyperphosphorylation induced by intracerebroventricular streptozotocin in rats. NeuroToxicology 2013, 38, 136–145.
- Kang, S.-W.; Kim, S.J.; Kim, M.-S. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59.
- Melov, S.; Adlard, P.A.; Morten, K.; Johnson, F.; Golden, T.R.; Hinerfeld, D.; Schilling, B.; Mavros, C.; Masters, C.L.; Volitakis, I.; et al. Mitochondrial Oxidative Stress Causes Hyperphosphorylation of Tau. PLoS ONE 2007, 2, e536.
- Chen, S.; Liu, A.-R.; An, F.-M.; Yao, W.-B.; Gao, X.-D. Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. AGE 2011, 34, 1211–1224.
- Clausen, A.; Xu, X.; Bi, X.; Baudry, M. Effects of the Superoxide Dismutase/Catalase Mimetic EUK-207 in a Mouse Model of Alzheimer’s Disease: Protection Against and Interruption of Progression of Amyloid and Tau Pathology and Cognitive Decline. J. Alzheimer’s Dis. 2012, 30, 183–208.
- Sanderson, T.H.; Reynolds, C.; Kumar, R.; Przyklenk, K.; Hüttemann, M. Molecular Mechanisms of Ischemia–Reperfusion Injury in Brain: Pivotal Role of the Mitochondrial Membrane Potential in Reactive Oxygen Species Generation. Mol. Neurobiol. 2012, 47, 9–23.
- Du Boff, B.; Götz, J.; Feany, M.B. Tau promotes neurodegeneration via DRP1 mislocalization in vivo. Neuron 2012, 75, 618–632.
- Wang, W.; Wang, X.; Fujioka, H.; Hoppel, C.; Whone, A.; Caldwell, M.; Cullen, P.; Liu, J.; Zhu, X. Parkinson’s disease–associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2015, 22, 54–63.
- Kandimalla, R.; Manczak, M.; Fry, D.; Suneetha, Y.; Sesaki, H.; Reddy, P.H. Reduced dynamin-related protein 1 protects against phosphorylated Tau-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 4881–4897.
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.; Peters, A.; Luebke, J.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T. Tau Accumulation Causes Mitochondrial Distribution Deficits in Neurons in a Mouse Model of Tauopathy and in Human Alzheimer’s Disease Brain. Am. J. Pathol. 2011, 179, 2071–2082.
- Schiefecker, A.J.; Putzer, G.; Braun, P.; Martini, J.; Strapazzon, G.; Antunes, A.P.; Mulino, M.; Pinggera, D.; Glodny, B.; Brugger, H.; et al. Total TauProtein as Investigated by Cerebral Microdialysis Increases in Hypothermic Cardiac Arrest: A Pig Study. Ther. Hypothermia Temp. Manag. 2021, 11, 28–34.
- Chen, Y.-J.; Nguyen, H.M.; Maezawa, I.; Grössinger, E.M.; Garing, A.L.; Kohler, R.; Jin, L.-W.; Wulff, H. The potassium channel KCa3.1 constitutes a pharmacological target for neuroinflammation associated with ischemia/reperfusion stroke. Br. J. Pharmacol. 2016, 36, 2146–2161.
- Wang, Z.; Tan, L.; Yu, J.-T. Axonal Transport Defects in Alzheimer’s Disease. Mol. Neurobiol. 2014, 51, 1309–1321.
- Li, X.-C.; Xia-Chun, L.; Wang, Z.-H.; Luo, Y.; Zhang, Y.; Liu, X.-P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.-P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 24756.
- Ittner, L.M.; Ke, Y.; Götz, J. Phosphorylated Tau Interacts with c-Jun N-terminal Kinase-interacting Protein 1 (JIP1) in Alzheimer Disease. J. Biol. Chem. 2009, 284, 20909–20916.
- Kanaan, N.; Morfini, G.A.; Lapointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J. Neurosci. 2011, 31, 9858–9868.
- Ibáñez-Salazar, A.; Bañuelos-Hernandez, B.; Rodriguez-Leyva, I.; Chi-Ahumada, E.; Monreal-Escalante, E.; Jiménez-Capdeville, M.E.; Rosales-Mendoza, S. Oxidative Stress Modifies the Levels and Phosphorylation State of Tau Protein in Human Fibroblasts. Front. Neurosci. 2017, 11, 495.
- Boland, B.; Kumar, A.; Lee, S.; Platt, F.M.; Wegiel, J.; Yu, W.H.; Nixon, R.A. Autophagy induction and autophagosome clearance in neurons: Relationship to autophagic pathology in Alzheimer’s disease. J. Neurosci. 2008, 28, 6926–6937.
- Vidal, R.L.; Matus, S.; Bargsted, L.; Hetz, C. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol. Sci. 2014, 35, 583–591.
- Maday, S.; Holzbaur, E.L.F. Compartment-Specific Regulation of Autophagy in Primary Neurons. J. Neurosci. 2016, 36, 5933–5945.
- Feng, J.; Chen, X.; Shen, J. Reactive nitrogen species as therapeutic targets for autophagy: Implication for ischemic stroke. Expert Opin. Ther. Targets 2017, 21, 305–317.
- Koike, M.A.; Green, K.N.; Blurton-Jones, M.; Laferla, F.M. Oligemic hypoperfusion differentially affects tau and amyloid-beta. Am. J Pathol. 2010, 177, 300–310.
- Huuskonen, M.T.; Loppi, S.; Dhungana, H.; Keksa-Goldsteine, V.; Lemarchant, S.; Korhonen, P.; Wojciechowski, S.; Pollari, E.; Valonen, P.; Koponen, J.; et al. Bexarotene targets autophagy and is protective against thromboembolic stroke in aged mice with tauopathy. Sci. Rep. 2016, 6, 33176.
- Falcon, B.; Noad, J.; McMahon, H.; Randow, F.; Goedert, M. Galectin-8–mediated selective autophagy protects against seeded tau aggregation. J. Biol. Chem. 2018, 293, 2438–2451.
- Scott, I.S.; Lowe, J.S. The ubiquitin-binding protein p62 identifies argyrophilic grain pathology with greater sensitivity than conventional silver stains. Acta Neuropathol. 2006, 113, 417–420.
- Ozcelik, S.; Fraser, G.; Castets, P.; Schaeffer, V.; Skachokova, Z.; Breu, K.; Clavaguera, F.; Sinnreich, M.; Kappos, L.; Goedert, M.; et al. Rapamycin Attenuates the Progression of Tau Pathology in P301S Tau Transgenic Mice. PLoS ONE 2013, 8, e62459.
- Xu, Y.; Martini-Stoica, H.; Zheng, H. A seeding based cellular assay of tauopathy. Mol. Neurodegener. 2016, 11, 1–10.
- Hasegawa, M. Molecular Mechanisms in the Pathogenesis of Alzheimer’s disease and Tauopathies-Prion-Like Seeded Aggregation and Phosphorylation. Biomolecules 2016, 6, 24.
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2014, 41, 24–46.
- Gusev, G.P.; Govekar, R.; Gadewal, N.; Agalakova, N.I. Understanding quasi-apoptosis of the most numerous enucleated components of blood needs detailed molecular autopsy. Ageing Res. Rev. 2017, 35, 46–62.
- Ma, X.; Liu, L.; Meng, J. MicroRNA-125b promotes neurons cell apoptosis and Tau phosphorylation in Alzheimer’s disease. Neurosci. Lett. 2017, 661, 57–62.
- Cheng, W.; Chen, W.; Wang, P.; Chu, J. Asiatic acid protects differentiated PC12 cells from Aβ25–35-induced apoptosis and tau hyperphosphorylation via regulating PI3K/Akt/GSK-3β signaling. Life Sci. 2018, 208, 96–101.
- Xiao, N.; Zhang, F.; Zhu, B.; Liu, C.; Lin, Z.; Wang, H.; Xie, W.-B. CDK5-mediated tau accumulation triggers methamphetamine-induced neuronal apoptosis via endoplasmic reticulum-associated degradation pathway. Toxicol. Lett. 2018, 292, 97–107.
- Kovac, A.; Zilka, N.; Kazmerova, Z.; Cente, M.; Zilkova, M.; Novak, M. Misfolded Truncated Protein τ Induces Innate Immune Response via MAPK Pathway. J. Immunol. 2011, 187, 2732–2739.
- Zilka, N.; Kazmerova, Z.; Jadhav, S.; Neradil, P.; Madari, A.; Obetkova, D.; Bugos, O.; Novak, M. Who fans the flames of Alz-heimer’s disease brains? Misfolded tau on the crossroad of neurodegenerative and inflammatory pathways. J. Neuroinflam. 2012, 9, 47.
- Asai, H.; Ikezu, S.; Woodbury, M.E.; Yonemoto, G.M.; Cui, L.; Ikezu, T. Accelerated Neurodegeneration and Neuroinflammation in Transgenic Mice Expressing P301L Tau Mutant and Tau-Tubulin Kinase 1. Am. J. Pathol. 2014, 184, 808–818.
- Majerova, P.; Zilkova, M.; Kazmerova, Z.; Kovac, A.; Paholikova, K.; Kovacech, B.; Zilka, N.; Novak, M. Microglia display modest phagocytic capacity for extracellular tau oligomers. J. Neuroinflam. 2014, 11, 1–12.
- Li, Y.; Liu, L.; Barger, S.; Griffin, W.S.T. Interleukin-1 Mediates Pathological Effects of Microglia on Tau Phosphorylation and on Synaptophysin Synthesis in Cortical Neurons through a p38-MAPK Pathway. J. Neurosci. 2003, 23, 1605–1611.
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421.
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; LaFerla, F.M. Lipopolysaccharide-induced inflammation exacer-bates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853.
- Janelsins, M.C.; Mastrangelo, M.A.; Park, K.M.; Sudol, K.L.; Narrow, W.C.; Oddo, S.; LaFerla, F.M.; Callahan, L.M.; Federoff, H.J.; Bowers, W.J. Chronic neuron-specific tumor necrosis factor-alpha expression enhances the local inflammatory environment ultimately leading to neuronal death in 3xTg-AD mice. Am. J. Pathol. 2008, 173, 1768–1782.
- Sy, M.; Kitazawa, M.; Medeiros, R.; Whitman, L.; Cheng, D.; Lane, T.E.; LaFerla, F.M. Inflammation Induced by Infection Potentiates Tau Pathological Features in Transgenic Mice. Am. J. Pathol. 2011, 178, 2811–2822.
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Loss of tau rescues inflammation-mediated neurodegeneration. Front. Neurosci. 2015, 9, 196.
- Kovac, A.; Zilkova, M.; Deli, M.A.; Zilka, N.; Novak, M. Human truncated tau is using a different mechanism from amyloid-beta to damage the blood-brain barrier. J. Alzheimer’s Dis. 2009, 18, 897–906.
- Mastrangelo, M.A.; Sudol, K.L.; Narrow, W.C.; Bowers, W.J. Interferon-gamma differentially affects Alzheimer’s disease pathologies and induces neurogenesis in triple transgenic-AD mice. Am. J. Pathol. 2009, 175, 2076–2088.
- Bolós, M.; Llorens-Martín, M.; Jurado-Arjona, J.; Hernández, F.; Rábano, A.; Avila, J. Direct Evidence of Internalization of Tau by Microglia In Vitro and In Vivo. J. Alzheimer’s Dis. 2015, 50, 77–87.

