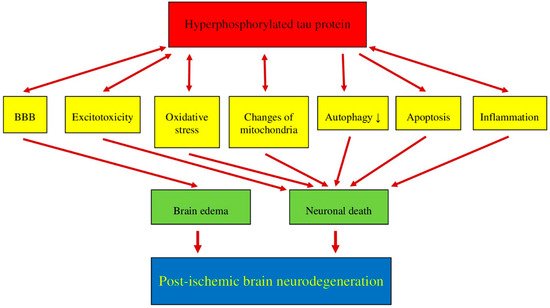
Recent data suggest that post-ischemic brain neurodegeneration in humans and animals is associated with the modified tau protein in a manner typical of Alzheimer’s disease neuropathology. Pathological changes in the tau protein, at the gene and protein level due to cerebral ischemia, can lead to the development of Alzheimer’s disease-type neuropathology and dementia. Some studies have shown increased tau protein staining and gene expression in neurons following ischemia-reperfusion brain injury. Recent studies have found the tau protein to be associated with oxidative stress, apoptosis, autophagy, excitotoxicity, neuroinflammation, blood-brain barrier permeability, mitochondrial dysfunction, and impaired neuronal function. In this review, we discuss the interrelationship of these phenomena with post-ischemic changes in the tau protein in the brain. The tau protein may be at the intersection of many pathological mechanisms due to severe neuropathological changes in the brain following ischemia. The data indicate that an episode of cerebral ischemia activates the damage and death of neurons in the hippocampus in a tau protein-dependent manner, thus determining a novel and important mechanism for the survival and/or death of neuronal cells following ischemia. In this review, we update our understanding of proteomic and genomic changes in the tau protein in post-ischemic brain injury and present the relationship between the modified tau protein and post-ischemic neuropathology and present a positive correlation between the modified tau protein and a post-ischemic neuropathology that has characteristics of Alzheimer’s disease-type neurodegeneration.
- brain ischemia
- protein
1. Post-Ischemic Tau Protein versus Blood-Brain Barrier