 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kha-Liang Lee | + 4022 word(s) | 4022 | 2020-08-06 10:54:36 | | | |
| 2 | Conner Chen | -40 word(s) | 3982 | 2020-10-30 07:32:15 | | |
Video Upload Options
A higher propensity of developing brain metastasis exists in triple-negative breast cancer (TNBC). Upon comparing the metastatic patterns of all breast cancer subtypes, patients with TNBC exhibited increased risks of the brain being the initial metastatic site, early brain metastasis development, and shortest brain metastasis-related survival. Notably, the development of brain metastasis differs from that at other sites owing to the brain-unique microvasculature (blood brain barrier (BBB)) and intracerebral microenvironment. Studies of brain metastases from TNBC have revealed the poorest treatment response, mostly because of the relatively backward strategies to target vast disease heterogeneity and poor brain efficacy. Moreover, TNBC is highly associated with the existence of cancer stem cells (CSCs), which contribute to circulating cancer cell survival before BBB extravasation, evasion from immune surveillance, and plasticity in adaptation to the brain-specific microenvironment. We summarized recent literature regarding molecules and pathways and reviewed the effects of CSC biology during the formation of brain metastasis in TNBC.
1. Introduction
After lung cancer, breast cancer is the second most frequently diagnosed origin of brain metastases [1]. Globally, breast cancer is the primary cause of cancer-related death in women and affects two million women annually, with more than 600,000 cases of mortality related to recurrence and metastases [2][3]. During the course of metastatic breast cancer, the central nervous system (CNS) is often involved later, with the incidence ranging from 25% to 46% [4]. Because of the improvement of early detection imaging and medications used for adjuvant and systemic therapy that have low blood brain barrier (BBB) penetrance, the actual and reported incidence of brain metastases in breast cancer is likely to increase. Risk factors for brain metastasis include age <35 years, higher histological grade, higher number of non-CNS metastatic sites, and the duration from the date of primary diagnosis [5]. Breast graded prognostic assessment (GPA) and modified breast-GPA have been noted to accurately predict overall survival (OS) in patients with breast cancer with brain metastases (p < 0.001 for both scores) [6] and are therefore, generally used in a clinical setting. The prognosticators of OS include age, the extent of primary disease control, the presence of extracranial metastases or leptomeningeal disease, Karnofsky Performance Status (KPS), and the availability of systemic treatment options [7]. Notably, the propensity to develop brain metastasis in advanced-stage breast cancer varies based on cancer subtypes [4][8][9].
2. Triple-Negative Breast Cancer and Brain Metastasis
Triple negative breast cancer (TNBC) accounts for 15% to 20% of breast cancers [10]. The diagnosis of the triple-negative subtype is made by excluding the expression or amplification of three biomarkers (the estrogen receptor (ER), the progesterone receptor (PR), and the human epidermal growth factor receptor 2 (HER2) protein), which are the oncogenic drivers and targets for breast cancer treatment. The disease typically presents as histologically high-grade-infiltrating ductal carcinoma [11], which mostly affects in younger women (age <40 years) [12]. Unlike the cancer subtypes involving the hormone receptor or HER2 markers that govern the choice of target therapy, the main aim of systemic treatment is to disrupt cancer cell survival in the TNBC subtype through chemotherapy regimens involving anthracyclines, alkylates, taxanes, and/or platinum [13][14][15]. Studies have proposed various TNBC classifications based on the identification of the following: (1) genomic expression, (2) histopathology, and (3) copy number and mutational analysis, in the hope of developing treatment-relevant classifications as a guide to treatment efficacy [16][17][18][19]. However, current researches have produced mixed results with varying conclusions. To date, patients with TNBC have the poorest prognosis, with the median progression-free survival (PFS) ranging from 3 to 4 months after the failure of first-line therapy, disease recurrence in one-half of early-stage patients and up to 37% of 5 year mortality rate after initial surgery [20][21].
The incidence of brain metastasis in advanced-stage breast cancer varies based on subtypes, with 30% to 46% of brain metastasis cases occurring in the triple-negative subtype, approximately one-third of the cases in the HER2-enriched subtype, and 14% of the cases in the luminal subtype [4][8][9]. Although most brain metastases occur at the advanced stages of cancer progression, TNBC usually spreads to the brain rapidly at earlier stages [11][22][23]. A 15 year cohort study reviewed the metastatic behavior of all breast cancer subtypes and observed that bone was the most common site of metastases for all early-stage breast cancer subtypes, except TNBC. Patients with basal-like TNBC had a higher rate of brain (odds ratio (OR), 3.7; 95% confidence interval (CI), 2.1–6.5), lung (OR, 2.5; 95% CI, 1.6–3.8), and distant nodal metastases (OR, 2.8; 95% CI, 1.8–4.5) but a significantly lower rate of liver (OR, 0.5; 95% CI, 0.3–0.8) and bone metastases (OR, 0.4; 95% CI, 0.2–0.6) compared with patients with the luminal cancer subtype. A similar pattern was found for non-basal triple-negative tumors, but they were not associated with fewer liver metastases [24]. Although the basal subtype is typically responsible for the aggressive behavior of TNBC in patients [25], no statistically significant difference was noted between the basal and non-basal biological subtypes regarding survival with brain metastases [26].
The incidence of brain metastasis in TNBC (BM-TNBC) varies significantly based on the disease stage. For instance, the 5 year cumulative incidence of the brain being the initial site of metastasis is 3%, 5%, and 10% for I, II, and III disease stages, respectively [27]. A case series reported that more than a quarter of BM-TNBC patients had brain metastasis as the first recurrence site [28]. In addition, patients with TNBC had the shortest interval (22 months) from primary early breast cancer to brain metastasis development compared with the luminal (63.5 months) or HER2-enriched (30 months) subtypes [22]. Furthermore, patients with BM-TNBC had a shorter median survival after brain metastasis development compared with the other subtypes (5–7 months vs. 10–18 months, respectively) [23][28][29]. A series comprised 433 patients with TNBC with variable metastatic sites and observed that median survival following a diagnosis of brain metastasis was 7.3 months. A longer median survival from the time of first recurrent brain metastasis was noted compared with those of subsequent recurrent (17.3 vs. 6.3 months, p = 0.008). However, patients with first recurrent brain metastasis were associated with shorter OS compared with those without brain metastasis (17.3 vs. 22.1 months, p = 0.006) [28]. In addition, a recent multicenter analysis of 219 patients with breast cancer with brain metastases reported the subtype switching between primary disease and brain metastases. The prevalence of any receptor discordance was 36.3%, and the receptor-specific discordance was 16.7% in ER, 25.2% in PR, and 10.4% in HER2 expression [30]. Notably, the discordance in receptor expression could be related to varying prognosis [31][32]. Therefore, a confirmatory biopsy should always be considered.
3. Understanding the Biology of Brain Metastases in Triple-Negative Breast Cancer
Cancer cells must invade through the extracellular matrix (ECM), intravasate blood vessels, survive the turbid flow of the vasculature, escape from immune surveillance, arrest in the capillary bed, extravasate, and finally colonize a distant metastatic site [33][34]. Patients with TNBC have a shorter interval to brain metastasis development and the shortest CNS-related survival among all breast cancer subtypes, suggesting the innate ability of TNBC tumor cells to adapt to the brain. The development of brain metastases occurs through the seeding of circulating tumor initiating cells (cancer stem cells (CSC)) into the brain microvasculature. In addition to the sanctuary site of the brain where the functional barriers resist the penetration of systemic medical therapies, tumor growth is aided by the unique intracerebral microenvironment [35]. Because of the genetic predisposition and cellular adaptation mechanisms, crosstalk between CSCs and brain-resident cells has rendered the molecular makeup of brain metastasis different from that of the primary tumors as well as from the metastases at other sites. In addition, we previously reported that TNBC tumors exhibit more traits of CSCs than the other breast cancer subtypes [36]. Herein, we discuss the mechanisms of BM-TNBC, namely BBB extravasation and brain-specific microenvironment interplay in relation to the CSC biology (Figure 1). Apart from the identification of drug targets defined by intracerebral tumors, identifying targets in CSCs and understanding the molecular mechanisms that support circulating CSCs to succeed in BBB extravasation, intracerebral seeding, and outgrowth in the brain are crucial in developing therapeutic strategies.
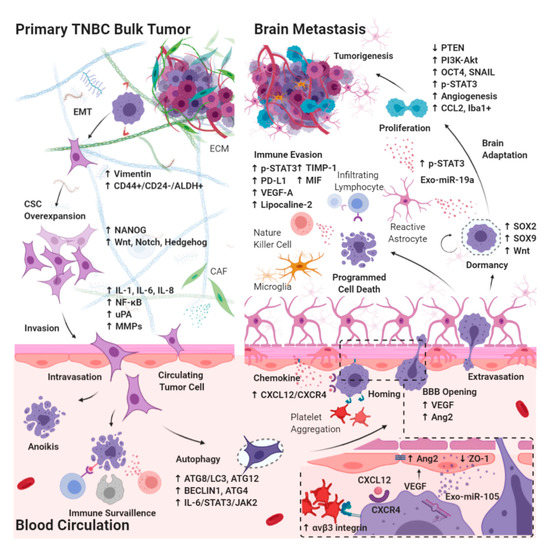
Figure 1. Diagram of molecular pathways involving CSC during the development of BM-TNBC. CSCs in TNBC bulk tumor must invade through the extracellular matrix in primary bulk tumors, intravasate blood vessels, survive the turbid flow of the blood circulation in vasculature, escape from immune surveillance, arrest in the capillary bed, and extravasate blood brain barrier (BBB), with subsequent tumor growth into brain metastasis involving microenvironmental niche–dormant cell interactions, neuroinflammatory cascades, and neovascularization. EMT: epithelial–mesenchymal transition; CAF: cancer-associated fibroblast; IL: interleukin; ATG: autophagy-related protein; VEGF: vascular endothelial growth factor; Exo-miR: microRNA enriched exosome; STAT: signal transducer and activator of transcription; JAK: janus kinase; uPA: urokinase plasminogen activator; MMP: matrix metalloproteinase; PD-L1: programmed death-1ligand 1; CXCL12: stromal cell-derived factor 1; CXCR4: C-X-C chemokine receptor type 4; Ang2: angiopoietin2; ZO: zonula occludens; SOX: SRY-box transcription factor; OCT4: octamer-binding transcription factor 4; PTEN: phosphatase and tensin homolog; TIMP: metallopeptidase inhibitor; MIF: macrophage migration inhibitory factor; SNAIL: zinc finger protein SNAI1; CCL2: monocyte chemoattractant protein-1; iba1+: ionized calcium binding adaptor myeloid cells.
3.1. Effects of CSC Biology on BM-TNBC
CSCs are multipotent stem cells responsible for the long-term clonal maintenance and growth of most human neoplasms. The coexistence of both the cycling and quiescent states of CSCs when they mutate or interact with their microenvironment further increases their heterogeneity and plasticity in overcoming the environmental stressors [37]. Among solid tumors, breast CSCs (BCSCs) were the first to be identified through the analysis of the expression of CD44/CD24 and ALDH1 in relation to the distinct levels of differentiation in the breast cancer population [38][39]. Their expressions are associated with therapy resistance, local recurrence, and distant metastasis, which clinically contribute to the poor therapeutic response, disease-free survival (DFS), PFS, and OS [40][41]. TNBC is highly associated with the existence of CSCs. For example, a TNBC cell line with mesenchymal origin, MDA-MB-231, is mostly composed (>90%) of cells that display the CD44+/CD24-/low BCSC immuno-phenotype [42][43]. When correlating with a perspective of CSC hypothesis in patients with TNBC, two independent biological levels, namely cellular and immune, are identified to stratify the prognostic and possible therapeutic classification [19]. Enriched ALDH1-expressing cells are an independent prognostic factor that predicts poor prognosis in patients with TNBC [44][45][46]. In addition, Notch, Hedgehog, and Wnt signaling pathways that regulate the maintenance of a stem cell niche, invasion capacity, and apoptosis escape also contribute to the therapy resistance phenotype of BCSCs during systemic chemotherapy treatment [47][48][49]. Notably, the activation of Wnt/β-catenin signaling, which is associated with the microglial-promoted colonization of breast cancer cells in the brain tissue [50].
3.1.1. Epithelial–Mesenchymal Transition in CSCs Breaking Away from the Primary Bulk Tumor
Evidence suggests that in various cancers, a subpopulation of circulating tumor cells (CTCs) has the CSC phenotype during the initial metastasis process [51][52][53] through the activation of epithelial–mesenchymal transition (EMT), which endows them with inherent metastatic potential [54]. The activation of EMT facilitates the invasion and migration of cancer cells from the primary tumor site to intravasation and translocation to distant metastasis sites [55], whereas the activation of mesenchymal epithelial transition (MET) after extravasation and cell seeding may facilitate colonization [56]. EMT transcriptional factors and the mesenchymal marker vimentin are expressed at higher levels in CD44+/CD24-/low BCSC-like cells than in more differentiated epithelial CD44-/CD24+ cells [57]. In addition, the changes in ECM dynamics may contribute to the disruption of asymmetric stem cell division, leading to CSC overexpansion [58]. Hyaluronic acid, which constitutes the ECM structures, interacts with the cell surface protein CD44, enhancing CSC properties by activating the stem cell marker NANOG [59]. CSCs in TNBC cell lines display highly invasive properties with elevated expression of proinvasive genes, such as those for interleukin (IL)-1, IL-6, IL-8, and urokinase plasminogen activator (uPA) [42]. Together with the stimulation of the nuclear factor kappa-light-chain enhancer of activated B cell (NF-κB) signaling, stromal uPA activates matrix metalloproteinase-2 (MMP-2) production and becomes responsive on the surface of tumor cells to cleave any component of the ECM, thereby allowing a break through the basement membrane and facilitating the EMT process [60][61].
3.1.2. Autophagy in Circulating CSCs Undergoing Circulatory Arrest and Evading Immune Surveillance
In patients with cancer, approximately 1 × 106 cancer cells per 1 g of tumor enter the circulation daily. However, only a fraction of these cells survive and reach a distant niche [62]. It has been reported that tumor cells take a longer time to extravasate the brain because of the extra boundaries contributed by the BBB [63]. In lung cancer, it takes approximately 48 h to extravasate the brain, whereas it takes only 6 h to extravasate the liver. On the other hand, breast cancer cells require 2 to 7 days to extravasate the brain. Consequently, CTCs have a longer survival time within the cerebral vasculature compared with that in other metastatic sites [64]. Studies have revealed that the mutation or inactivation of cell cycle-regulating genes and apoptosis-inducing genes enable CSCs to escape apoptosis [65]. In addition to DNA repair systems, autophagy is one of the processes that is strongly associated with CSC physiology in TNBC cells, including tumorigenesis, differentiation, plasticity, migration, or invasion, as well as pharmacological, viral, and immunoresistance [66][67][68]. Autophagy is a cellular degradation process that is essential for promoting cellular adaptation to stressors, cytokine mediation, chromosome stability, stem cell microenvironment, and cellular homeostatic mechanism [66][69][70][71][72]. It has been determined that in triple-negative BCSC, the knock down of autophagy-related genes, such as ATG8/LC3 and ATG12, or pharmacological blockade of autophagy flux decreases the CD44+/CD24-/low CSC phenotype [73], probably through the IL6/STAT3/JAK2 pathways that contribute to the conversion of non-CSCs into CSCs [65]. The autophagy process can be induced upon the loss of integrin-mediated cell attachment to the surrounding ECM [74], and it further protects cells from detachment-induced cell death, termed anoikis [75]. Two key autophagy proteins, BECLIN1 and ATG4, are upregulated in mammospheres compared with adherent cells and are needed for their maintenance and expansion [76]. Upon EMT and autophagy induction, the process aids in the evasion from immune surveillance programs, such as T-cell-mediated lysis [77][78] and inhibit NK-mediated tumor cell killing by degrading granzyme B [79][80].
3.2. Extravasation from the BBB
The BBB is a complex functional boundary that tightly regulates the transmission of molecular and cellular substances into the brain. It is made up of the large hydrophilic molecules of endothelial cells, pericytes, astrocytic endfeets, and neuronal cells, which have evolved into protective insulation for neuronal signaling. Junctional complex proteins include claudins, occludins, and zona occluden (ZO) proteins, and junctional adhesion molecules connect endothelial cells with continuous tight junctions that limit the transcytosis rate. Two basement membranes (endothelial and astrocytic), namely embedded pericytes and astrocytic endfeets, play a crucial role in the maintenance and regulation of BBB permeability. Mechanistically, pericytes regulate BBB permeability through the expressions of transporters and the physical constriction of the blood vessels, whereas astrocytes cover most of the BBB surface area functioning to couple the endothelial cells and pericytes [81][82][83].
3.2.1. Chemokines Ligand Receptor System on Circulating CSCs Migrating through the BBB
Therefore, to enter the brain parenchyma, cancer cells must pass through microcapillary walls by opening tight junctions [84]. These junctions can be destabilized by cancer cells through the expression of cytokines, chemokines, and inflammatory mediators [85][86]. However, it is not yet clear how circulating CSCs affect the function of the BBB. Current potential mechanisms of the extravasation strategy of tumor cells include docking, locking, trans-endothelial migration, and adhesion to the subendothelial matrix, which mimic the mechanisms used by immune cells [87]. The CXCL12/CXCR4/CXCR7 ligand receptor system is the vital axis that regulates neuro-glio-vascular interactions and vessel growth during human brain development. In a traumatic brain injury murine model, the CXCL12/CXCR4 axis was stimulated to promote neural stem cell migration through the stimulation of MMP-2 secretion [88][89]. Nevertheless, the CXCL12/CXCR4 axis promotes metastasis and invasion across a wide variety of solid tumors. For example, the CXCL12/CXCR4 autocrine-positive feedback mechanism controls the survival and proliferation of neural progenitor derived glioblastomas under hypoxic stress [90][91][92]. In breast cancer brain metastases, the CXCL12/CXCR4 axis takes part in the homing, motility, and progression of metastases to regulate the migration of CTCs through the BBB. Notably, BCSCs have high CXCR4 expression and are defined as a pro-metastatic subpopulation, whereas the expression of CXCL12 defines regions with a higher risk of causing metastasis for breast cancer invasion [93][94].
3.2.2. Interaction between Brain Microvascular Endothelial Cells and Circulating CSCs Mediates the Tight Junction Disruption and BBB Destabilization
Moreover, cancer cell-derived exosomes have been noted to have multiple roles in the events of metastasis, with a key role in pre-metastatic niche formation through vascular remodeling and modulation of cellular behaviors in the pre-metastatic site. Metastatic TNBC cells secrete miR-105-enriched exosomes to down-regulate ZO-1 protein expression in endothelial cells, disrupting the endothelial cell barrier and increasing vascular permeability, thereby facilitating invasion and migration [95]. Reciprocally, brain endothelial cells actively influence tumor cell extravasation and proliferation, which are mediated by bilateral cell surface receptors and adhesion molecules, such as integrins, selectins, chemokines, and the receptors of the immunoglobulin superfamily (ICAM-1, VCAM1) [96]. Activated αvβ3 integrin in circulating TNBC cells appeared to interact with platelets causing thrombus formation, which facilitates the arrest of tumor cells within the vasculature [97]. Furthermore, the activation of αvβ3-integrin assists in the intracerebral growth of TNBC cells through the continued upregulation of vascular endothelial growth factor (VEGF) [98]. An in vivo study injected brain-seeking TNBC cell lines into the murine carotid artery and demonstrated that the VEGF secreted from the cells destabilized the brain microvascular endothelial cells through activation of angiopoietin-2 (Ang2) expression, which further mediated BBB disruption (changes in the tight junction between ZO-1 and claudin-5). The interaction plays a critical role in the initial cascade of colonization of TNBC in the brain tissue, because the administration of neutralizing Ang-2 peptibody prevented changes to the BBB integrity and further inhibited the formation of TNBC cells colonization into the brain [99].
3.3. Intracerebral Tumor Microenvironment
3.3.1. Dormant Period of CSCs Adaptation to the Brain Microenvironment
Evidence supports dynamic crosstalk between cancer cells and the brain microenvironment contributed by surrounding stromal cells, immune cells, and ECM, which are crucial for tumor growth after cell seeding in the brain [100]. Nonetheless, most CTCs are likely to die even after successfully arresting and extravasating the brain [101]. The adaptation period following dissemination is typically preceded by a period of dormancy in which cells are in a slow cell-cycling state; this is mediated by the expressions of stemness-associated transcription factors such as the SOX2, SOX9, and Wnt signaling pathways [102], which can last up to several decades [103]. Further exit from dormancy requires evasion from the immune surveillance mechanism and angiogenic switch to form micro-metastases [104]. Striking overlap exists between the behaviors of CSCs in tumors and the behavior of cancer cells in a dormant state, especially in the context of tumorigenesis after cell seeding; this suggests that the dormant extravasated tumor shows the CSC phenotype [105][106]. A study revealed that a combination of intracellular and extracellular signals within the tumor microenvironment, namely regulation of quiescence, alteration of angiogenic response, and modulation of immune surveillance govern the regulation of the dormant state [107].
3.3.2. Interaction between Reactive Astrocytes and CSCs Evading Immune Surveillance through the Activation of STAT3 Pathway
Astrocytes have long been recognized as a key stromal component and immunomodulator of metastatic brain tumors which have tumor-killing or tumor-promoting effects, likely reflecting the fact that these cells exist as distinct subtypes with distinct functions [108][109][110]. Several astrocyte subtypes have been identified, termed A1 and A2 [111]. A1 astrocytes are regarded as proinflammatory astrocytes, which are induced by classically activated neuroinflammatory microglia through the secretion of IL-1a, TNF, and C1q. In the event of CNS injury and neurodegenerative disease, A1 astrocytes lose the ability to promote neuronal survival, outgrowth, synaptogenesis, and phagocytosis, thereby inducing the death of neurons and oligodendrocytes [112]. A2 astrocytes are thought to promote tissue repair through the production of neurotrophic factors. It is thought that most tumor-associated astrocytes are likely to be of the A2 subtype, which highly expresses the phosphorylated (activated) form of the signal transducer and activator of transcription-3 (p-STAT3) pathway. Notably, STAT3 signaling in tumor cell is a key signaling mechanism activated by cytokines and growth factor receptors that establish an immunosuppressive microenvironment during the early stages of breast carcinogenesis to promote tumor growth and metastases [113]. In addition, STAT3 expression in the BCSC-like subset of the TNBC cell line serves as a chemo-resistant marker [114]. Mechanistically, these p-STAT3+ reactive astrocytes block the access of immune cells, such as CD8+ cytotoxic T cell, to cancer cells through the upregulation of immunosuppressive molecules, such as programmed cell death–1 ligand 1 (PD-L1), VEGF-A, lipocalin-2, and tissue inhibitor of metalloproteinases 1 (TIMP-1). Furthermore, the crosstalk between microglia and reactive astrocytes contributes to the establishment of an immunosuppressive microenvironment, thereby facilitating the metastasis of breast cancer to the brain. The p-STAT3+ reactive astrocytes exhibit increased expression of the CD74 ligand, MIF (macrophage migration inhibitory factor), and increased binding to CD74+ microglia, which upregulate the expression of midkine (neurite growth-promoting factor 2 (NEGF2), a downstream target of the NF–κB signaling pathway that promotes the development of brain metastases. All pieces of evidence suggest the significance of the STAT3 pathway during the interplay between the TNBC CSCs and astrocytes contributing to the microenvironment.
3.3.3. Tumor Progression through the Activation of PI3K/Akt Signaling by Interaction between Reactive Astrocytes and CSCs
In addition, the phosphoinositide 3-kinase (PI3K)-Akt pathway has been implicated as a regulator of brain metastasis. PI3K signaling promotes various cellular processes that include proliferation, survival, metabolism, and angiogenesis in response to extracellular signals that activate receptor tyrosine kinases or G-protein coupled receptors. Notably, a study revealed that PI3K signaling in human breast cancer brain metastasis samples was not only active in the metastatic cells but also in the CNS microenvironment. Furthermore, in vitro and in vivo demonstrations revealed that treatment with buparlisib (BKM120), a pan-PI3K class 1 inhibitor with excellent BBB penetrance, reduced the macrophage or microglia-induced invasion and glial-assisted infiltration of breast cancer cells into the brain parenchyma [115]. The Wnt pathway acts in a compensatory manner when the TNBC cell line is challenged with buparlisib. Effectively, the dual PI3K and Wnt pathway inhibitor works in synergy to promote cell viability and enhance antitumor efficacy in TNBC cell lines [116]. In addition, results demonstrate that rational combinations of the buparlisib with MEK1/2 inhibitor in mice bearing intracranial TNBC tumors (SUM149, MDA-MB-231Br, MDA-MB-468, or MDA-MB-436) improved survival for intracranial SUM149 and MDA-MB-231Br, but not MDA-MB-468 or MDA-MB-436, suggesting therapeutic potential for the treatment of some BM-TNBC [117].
3.3.4. Brain-Specific PTEN Suppression in Maintaining CSCs Plasticity during Tumorigenesis
Furthermore, TNBC cells manifest distinct gene expression patterns after metastasizing to different organs because the interplay with the site-specific extrinsic signals affects subsequent metastatic outgrowth. For example, mutations of phosphatase and tensin homolog (PTEN), a tumor suppressor gene and negative regulator of PI3K-Akt signaling, are rarely observed in the primary breast or lung cancers; however, a 21% increase in PTEN mutations was observed in brain metastatic tumors [118][119]. In a study with 111 tissue samples of breast cancer brain metastases, loss of PTEN protein expression was noted in 48.6% of all samples. Notably, it is significantly associated with the TNBC subtype (67.5%, p = 0.001) [120]. Previous studies have revealed that PTEN inhibition increased the expressions of the stemness and mesenchymal markers, OCT4 and SNAIL in TNBC cell lines [121], suggesting the role of PTEN suppression in CSC maintenance of brain metastasis. In addition, loss of PTEN expression in TNBC cells seemed to be the essential trait acquired from the brain-specific microenvironment, which led to an increased secretion of cytokine chemokine ligand 2 (CCL2, chemo-attractant during inflammation) and the recruitment of Iba1+ myeloid cells that enhance tumor cells proliferation and reduce apoptosis [122]. PTEN overexpression was evidenced to attenuate the invasiveness and migration of breast cancer cells as well as astrocyte activation. Mechanistically, in the brain-specific microenvironment, reactive astrocytes suppress PTEN expression in TNBC cells with the intercellular transfer of PTEN-targeting microRNA (miR-19a), through exosomes secretion, whereas the blockade of astrocyte exosome secretion rescues PTEN loss and suppresses brain metastasis in vivo . Notably, the PTEN level in TNBC cells is restored after the cells leave the brain microenvironment. All these studies suggest that the critical role of plasticity in PTEN suppression might contribute to the CSC phenotype in brain-adaptive TNBC cells, thereby indicating new opportunities for effective anti-metastatic therapies in BM-TNBC [123][124][125].
References
- Barnholtz-Sloan, J.S.; Sloan, A.E.; Davis, F.G.; Vigneau, F.D.; Lai, P.; Sawaya, R.E. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J. Clin. Oncol. 2004, 22, 2865–2872, doi:10.1200/JCO.2004.12.149.
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30, doi:10.3322/caac.21387.
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108, doi:10.3322/caac.21262.
- Lin, N.U.; Claus, E.; Sohl, J.; Razzak, A.R.; Arnaout, A.; Winer, E.P. Sites of distant recurrence and clinical outcomes in patients with metastatic triple-negative breast cancer: High incidence of central nervous system metastases. Cancer 2008, 113, 2638–2645, doi:10.1002/cncr.23930.
- Graesslin, O.; Abdulkarim, B.S.; Coutant, C.; Huguet, F.; Gabos, Z.; Hsu, L.; Marpeau, O.; Uzan, S.; Pusztai, L.; Strom, E.A.; et al. Nomogram to predict subsequent brain metastasis in patients with metastatic breast cancer. J. Clin. Oncol. 2010, 28, 2032–2037, doi:10.1200/JCO.2009.24.6314.
- Subbiah, I.M.; Lei, X.; Weinberg, J.S.; Sulman, E.P.; Chavez-MacGregor, M.; Tripathy, D.; Gupta, R.; Varma, A.; Chouhan, J.; Guevarra, R.P.; et al. Validation and Development of a Modified Breast Graded Prognostic Assessment As a Tool for Survival in Patients With Breast Cancer and Brain Metastases. J. Clin. Oncol. 2015, 33, 2239–2245, doi:10.1200/JCO.2014.58.8517.
- Ekici, K.; Temelli, O.; Dikilitas, M.; Halil Dursun, I.; Bozdag Kaplan, N.; Kekilli, E. Survival and prognostic factors in patients with brain metastasis: Single center experience. J. BUON 2016, 21, 958–963.
- Aversa, C.; Rossi, V.; Geuna, E.; Martinello, R.; Milani, A.; Redana, S.; Valabrega, G.; Aglietta, M.; Montemurro, F. Metastatic breast cancer subtypes and central nervous system metastases. Breast 2014, 23, 623–628, doi:10.1016/j.breast.2014.06.009.
- Pestalozzi, B.C.; Holmes, E.; de Azambuja, E.; Metzger-Filho, O.; Hogge, L.; Scullion, M.; Lang, I.; Wardley, A.; Lichinitser, M.; Sanchez, R.I.; et al. CNS relapses in patients with HER2-positive early breast cancer who have and have not received adjuvant trastuzumab: A retrospective substudy of the HERA trial (BIG 1-01). Lancet Oncol. 2013, 14, 244–248, doi:10.1016/S1470-2045(13)70017-2.
- Schmadeka, R.; Harmon, B.E.; Singh, M. Triple-negative breast carcinoma: Current and emerging concepts. Am. J. Clin. Pathol. 2014, 141, 462–477, doi:10.1309/AJCPQN8GZ8SILKGN.
- Qiu, J.; Xue, X.; Hu, C.; Xu, H.; Kou, D.; Li, R.; Li, M. Comparison of Clinicopathological Features and Prognosis in Triple-Negative and Non-Triple Negative Breast Cancer. J. Cancer 2016, 7, 167–173, doi:10.7150/jca.10944.
- Trivers, K.F.; Lund, M.J.; Porter, P.L.; Liff, J.M.; Flagg, E.W.; Coates, R.J.; Eley, J.W. The epidemiology of triple-negative breast cancer, including race. Cancer Causes Control. 2009, 20, 1071–1082, doi:10.1007/s10552-009-9331-1.
- Hayes, D.F.; Thor, A.D.; Dressler, L.G.; Weaver, D.; Edgerton, S.; Cowan, D.; Broadwater, G.; Goldstein, L.J.; Martino, S.; Ingle, J.N.; et al. HER2 and response to paclitaxel in node-positive breast cancer. N. Engl. J. Med. 2007, 357, 1496–1506, doi:10.1056/NEJMoa071167.
- Hugh, J.; Hanson, J.; Cheang, M.C.; Nielsen, T.O.; Perou, C.M.; Dumontet, C.; Reed, J.; Krajewska, M.; Treilleux, I.; Rupin, M.; et al. Breast cancer subtypes and response to docetaxel in node-positive breast cancer: Use of an immunohistochemical definition in the BCIRG 001 trial. J. Clin. Oncol. 2009, 27, 1168–1176, doi:10.1200/JCO.2008.18.1024.
- Martin, M.; Rodriguez-Lescure, A.; Ruiz, A.; Alba, E.; Calvo, L.; Ruiz-Borrego, M.; Santaballa, A.; Rodriguez, C.A.; Crespo, C.; Abad, M.; et al. Molecular predictors of efficacy of adjuvant weekly paclitaxel in early breast cancer. Breast Cancer Res. Treat. 2010, 123, 149–157, doi:10.1007/s10549-009-0663-z.
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767, doi:10.1172/JCI45014.
- Bareche, Y.; Venet, D.; Ignatiadis, M.; Aftimos, P.; Piccart, M.; Rothe, F.; Sotiriou, C. Unravelling triple-negative breast cancer molecular heterogeneity using an integrative multiomic analysis. Ann. Oncol. 2018, 29, 895–902, doi:10.1093/annonc/mdy024.
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.; Savage, M.I.; Osborne, C.K.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive genomic analysis identifies novel subtypes and targets of triple-negative breast cancer. Clin. Cancer Res. 2015, 21, 1688–1698, doi:10.1158/1078-0432.CCR-14-0432.
- Prado-Vazquez, G.; Gamez-Pozo, A.; Trilla-Fuertes, L.; Arevalillo, J.M.; Zapater-Moros, A.; Ferrer-Gomez, M.; Diaz-Almiron, M.; Lopez-Vacas, R.; Navarro, H.; Main, P.; et al. A novel approach to triple-negative breast cancer molecular classification reveals a luminal immune-positive subgroup with good prognoses. Sci. Rep. 2019, 9, 1538, doi:10.1038/s41598-018-38364-y.
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281, doi:10.1200/JCO.2007.14.4147.
- Mustacchi, G.; De Laurentiis, M. The role of taxanes in triple-negative breast cancer: Literature review. Drug Des. Devel. Ther. 2015, 9, 4303–4318, doi:10.2147/DDDT.S86105.
- Heitz, F.; Harter, P.; Lueck, H.J.; Fissler-Eckhoff, A.; Lorenz-Salehi, F.; Scheil-Bertram, S.; Traut, A.; du Bois, A. Triple-negative and HER2-overexpressing breast cancers exhibit an elevated risk and an earlier occurrence of cerebral metastases. Eur. J. Cancer 2009, 45, 2792–2798, doi:10.1016/j.ejca.2009.06.027.
- Niikura, N.; Hayashi, N.; Masuda, N.; Takashima, S.; Nakamura, R.; Watanabe, K.; Kanbayashi, C.; Ishida, M.; Hozumi, Y.; Tsuneizumi, M.; et al. Treatment outcomes and prognostic factors for patients with brain metastases from breast cancer of each subtype: A multicenter retrospective analysis. Breast Cancer Res. Treat. 2014, 147, 103–112, doi:10.1007/s10549-014-3090-8.
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277, doi:10.1200/JCO.2009.25.9820.
- Yamamoto, Y.; Ibusuki, M.; Nakano, M.; Kawasoe, T.; Hiki, R.; Iwase, H. Clinical significance of basal-like subtype in triple-negative breast cancer. Breast Cancer 2009, 16, 260–267, doi:10.1007/s12282-009-0150-8.
- Niwinska, A.; Olszewski, W.; Murawska, M.; Pogoda, K. Triple-negative breast cancer with brain metastases: A comparison between basal-like and non-basal-like biological subtypes. J. Neurooncol. 2011, 105, 547–553, doi:10.1007/s11060-011-0616-3.
- Dawood, S.; Lei, X.; Litton, J.K.; Buchholz, T.A.; Hortobagyi, G.N.; Gonzalez-Angulo, A.M. Incidence of brain metastases as a first site of recurrence among women with triple receptor-negative breast cancer. Cancer 2012, 118, 4652–4659, doi:10.1002/cncr.27434.
- Jin, J.; Gao, Y.; Zhang, J.; Wang, L.; Wang, B.; Cao, J.; Shao, Z.; Wang, Z. Incidence, pattern and prognosis of brain metastases in patients with metastatic triple negative breast cancer. BMC Cancer 2018, 18, 446, doi:10.1186/s12885-018-4371-0.
- Sperduto, P.W.; Kased, N.; Roberge, D.; Xu, Z.; Shanley, R.; Luo, X.; Sneed, P.K.; Chao, S.T.; Weil, R.J.; Suh, J.; et al. Effect of tumor subtype on survival and the graded prognostic assessment for patients with breast cancer and brain metastases. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 2111–2117, doi:10.1016/j.ijrobp.2011.02.027.
- Hulsbergen, A.F.C.; Claes, A.; Kavouridis, V.K.; Ansaripour, A.; Nogarede, C.; Hughes, M.E.; Smith, T.R.; Brastianos, P.K.; Verhoeff, J.J.C.; Lin, N.U.; et al. Subtype switching in breast cancer brain metastases: A multicenter analysis. Neuro Oncol. 2020, doi:10.1093/neuonc/noaa013.
- Kaidar-Person, O.; Meattini, I.; Jain, P.; Bult, P.; Simone, N.; Kindts, I.; Steffens, R.; Weltens, C.; Navarria, P.; Belkacemi, Y.; et al. Discrepancies between biomarkers of primary breast cancer and subsequent brain metastases: An international multicenter study. Breast Cancer Res. Treat. 2018, 167, 479–483, doi:10.1007/s10549-017-4526-8.
- Priedigkeit, N.; Hartmaier, R.J.; Chen, Y.; Vareslija, D.; Basudan, A.; Watters, R.J.; Thomas, R.; Leone, J.P.; Lucas, P.C.; Bhargava, R.; et al. Intrinsic Subtype Switching and Acquired ERBB2/HER2 Amplifications and Mutations in Breast Cancer Brain Metastases. JAMA Oncol. 2017, 3, 666–671, doi:10.1001/jamaoncol.2016.5630.
- Massague, J.; Obenauf, A.C. Metastatic colonization by circulating tumour cells. Nature 2016, 529, 298–306, doi:10.1038/nature17038.
- Talmadge, J.E.; Fidler, I.J. AACR centennial series: The biology of cancer metastasis: Historical perspective. Cancer Res. 2010, 70, 5649–5669, doi:10.1158/0008-5472.CAN-10-1040.
- Achrol, A.S.; Rennert, R.C.; Anders, C.; Soffietti, R.; Ahluwalia, M.S.; Nayak, L.; Peters, S.; Arvold, N.D.; Harsh, G.R.; Steeg, P.S.; et al. Brain metastases. Nat. Rev. Dis. Primers 2019, 5, 5, doi:10.1038/s41572-018-0055-y.
- Lee, K.L.; Kuo, Y.C.; Ho, Y.S.; Huang, Y.H. Triple-Negative Breast Cancer: Current Understanding and Future Therapeutic Breakthrough Targeting Cancer Stemness. Cancers 2019, 11, doi:10.3390/cancers11091334.
- Kreso, A.; Dick, J.E. Evolution of the cancer stem cell model. Cell Stem Cell 2014, 14, 275–291, doi:10.1016/j.stem.2014.02.006.
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988, doi:10.1073/pnas.0530291100.
- Ricardo, S.; Vieira, A.F.; Gerhard, R.; Leitao, D.; Pinto, R.; Cameselle-Teijeiro, J.F.; Milanezi, F.; Schmitt, F.; Paredes, J. Breast cancer stem cell markers CD44, CD24 and ALDH1: Expression distribution within intrinsic molecular subtype. J. Clin. Pathol. 2011, 64, 937–946, doi:10.1136/jcp.2011.090456.
- Chen, Y.; Song, J.; Jiang, Y.; Yu, C.; Ma, Z. Predictive value of CD44 and CD24 for prognosis and chemotherapy response in invasive breast ductal carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 11287–11295.
- Lin, Y.; Zhong, Y.; Guan, H.; Zhang, X.; Sun, Q. CD44+/CD24- phenotype contributes to malignant relapse following surgical resection and chemotherapy in patients with invasive ductal carcinoma. J. Exp. Clin. Cancer Res. 2012, 31, 59, doi:10.1186/1756-9966-31-59.
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R., Jr.; Badve, S.; Nakshatri, H. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006, 8, R59, doi:10.1186/bcr1610.
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68, doi:10.1186/bcr2635.
- Li, H.; Ma, F.; Wang, H.; Lin, C.; Fan, Y.; Zhang, X.; Qian, H.; Xu, B. Stem cell marker aldehyde dehydrogenase 1 (ALDH1)-expressing cells are enriched in triple-negative breast cancer. Int. J. Biol. Markers 2013, 28, e357–364, doi:10.5301/jbm.5000048.
- Ma, F.; Li, H.; Li, Y.; Ding, X.; Wang, H.; Fan, Y.; Lin, C.; Qian, H.; Xu, B. Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC). Medicine 2017, 96, e6561, doi:10.1097/MD.0000000000006561.
- Ohi, Y.; Umekita, Y.; Yoshioka, T.; Souda, M.; Rai, Y.; Sagara, Y.; Sagara, Y.; Sagara, Y.; Tanimoto, A. Aldehyde dehydrogenase 1 expression predicts poor prognosis in triple-negative breast cancer. Histopathology 2011, 59, 776–780, doi:10.1111/j.1365-2559.2011.03884.x.
- de Sousa, E.M.F.; Vermeulen, L. Wnt Signaling in Cancer Stem Cell Biology. Cancers 2016, 8, pii: E60, doi:10.3390/cancers8070060.
- Mamaeva, V.; Niemi, R.; Beck, M.; Ozliseli, E.; Desai, D.; Landor, S.; Gronroos, T.; Kronqvist, P.; Pettersen, I.K.; McCormack, E.; et al. Inhibiting Notch Activity in Breast Cancer Stem Cells by Glucose Functionalized Nanoparticles Carrying gamma-secretase Inhibitors. Mol. Ther. 2016, 24, 926–936, doi:10.1038/mt.2016.42.
- Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554–1585, doi:10.3390/cancers7030851.
- Pukrop, T.; Dehghani, F.; Chuang, H.N.; Lohaus, R.; Bayanga, K.; Heermann, S.; Regen, T.; Van Rossum, D.; Klemm, F.; Schulz, M.; et al. Microglia promote colonization of brain tissue by breast cancer cells in a Wnt-dependent way. Glia 2010, 58, 1477–1489, doi:10.1002/glia.21022.
- Aktas, B.; Tewes, M.; Fehm, T.; Hauch, S.; Kimmig, R.; Kasimir-Bauer, S. Stem cell and epithelial-mesenchymal transition markers are frequently overexpressed in circulating tumor cells of metastatic breast cancer patients. Breast Cancer Res. 2009, 11, R46, doi:10.1186/bcr2333.
- Theodoropoulos, P.A.; Polioudaki, H.; Agelaki, S.; Kallergi, G.; Saridaki, Z.; Mavroudis, D.; Georgoulias, V. Circulating tumor cells with a putative stem cell phenotype in peripheral blood of patients with breast cancer. Cancer Lett. 2010, 288, 99–106, doi:10.1016/j.canlet.2009.06.027.
- Kasimir-Bauer, S.; Hoffmann, O.; Wallwiener, D.; Kimmig, R.; Fehm, T. Expression of stem cell and epithelial-mesenchymal transition markers in primary breast cancer patients with circulating tumor cells. Breast Cancer Res. 2012, 14, R15, doi:10.1186/bcr3099.
- Yang, M.H.; Imrali, A.; Heeschen, C. Circulating cancer stem cells: The importance to select. Chin. J. Cancer Res. 2015, 27, 437–449, doi:10.3978/j.issn.1000-9604.2015.04.08.
- Li, F.; Tiede, B.; Massague, J.; Kang, Y. Beyond tumorigenesis: Cancer stem cells in metastasis. Cell Res. 2007, 17, 3–14, doi:10.1038/sj.cr.7310118.
- Tam, W.L.; Weinberg, R.A. The epigenetics of epithelial-mesenchymal plasticity in cancer. Nat. Med. 2013, 19, 1438–1449, doi:10.1038/nm.3336.
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715, doi:10.1016/j.cell.2008.03.027.
- Lu, P.; Weaver, V.M.; Werb, Z. The extracellular matrix: A dynamic niche in cancer progression. J. Cell Biol. 2012, 196, 395–406, doi:10.1083/jcb.201102147.
- Bourguignon, L.Y.; Peyrollier, K.; Xia, W.; Gilad, E. Hyaluronan-CD44 interaction activates stem cell marker Nanog, Stat-3-mediated MDR1 gene expression, and ankyrin-regulated multidrug efflux in breast and ovarian tumor cells. J. Biol. Chem. 2008, 283, 17635–17651, doi:10.1074/jbc.M800109200.
- Radisky, E.S.; Radisky, D.C. Matrix metalloproteinase-induced epithelial-mesenchymal transition in breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 201–212, doi:10.1007/s10911-010-9177-x.
- Weigelt, B.; Peterse, J.L.; van ‘‘t Veer, L.J. Breast cancer metastasis: Markers and models. Nat. Rev. Cancer 2005, 5, 591–602, doi:10.1038/nrc1670.
- Chang, Y.S.; di Tomaso, E.; McDonald, D.M.; Jones, R.; Jain, R.K.; Munn, L.L. Mosaic blood vessels in tumors: Frequency of cancer cells in contact with flowing blood. Proc. Natl. Acad. Sci. USA 2000, 97, 14608–14613, doi:10.1073/pnas.97.26.14608.
- Wrobel, J.K.; Toborek, M. Blood-brain Barrier Remodeling during Brain Metastasis Formation. Mol. Med. 2016, 22, 32–40, doi:10.2119/molmed.2015.00207.
- Kim, N.H.; Kim, H.S.; Li, X.Y.; Lee, I.; Choi, H.S.; Kang, S.E.; Cha, S.Y.; Ryu, J.K.; Yoon, D.; Fearon, E.R.; et al. A p53/miRNA-34 axis regulates Snail1-dependent cancer cell epithelial-mesenchymal transition. J. Cell Biol. 2011, 195, 417–433, doi:10.1083/jcb.201103097.
- Maycotte, P.; Jones, K.L.; Goodall, M.L.; Thorburn, J.; Thorburn, A. Autophagy Supports Breast Cancer Stem Cell Maintenance by Regulating IL6 Secretion. Mol. Cancer Res. 2015, 13, 651–658, doi:10.1158/1541-7786.MCR-14-0487.
- Nazio, F.; Bordi, M.; Cianfanelli, V.; Locatelli, F.; Cecconi, F. Autophagy and cancer stem cells: Molecular mechanisms and therapeutic applications. Cell Death Differ. 2019, 26, 690–702, doi:10.1038/s41418-019-0292-y.
- Yeo, S.K.; Wen, J.; Chen, S.; Guan, J.L. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfbeta/Smad Signaling. Cancer Res. 2016, 76, 3397–3410, doi:10.1158/0008-5472.CAN-15-2946.
- Robert, T.; Vanoli, F.; Chiolo, I.; Shubassi, G.; Bernstein, K.A.; Rothstein, R.; Botrugno, O.A.; Parazzoli, D.; Oldani, A.; Minucci, S.; et al. HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 2011, 471, 74–79, doi:10.1038/nature09803.
- Mathew, R.; Kongara, S.; Beaudoin, B.; Karp, C.M.; Bray, K.; Degenhardt, K.; Chen, G.; Jin, S.; White, E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev. 2007, 21, 1367–1381, doi:10.1101/gad.1545107.
- Janji, B.; Viry, E.; Moussay, E.; Paggetti, J.; Arakelian, T.; Mgrditchian, T.; Messai, Y.; Noman, M.Z.; Van Moer, K.; Hasmim, M.; et al. The multifaceted role of autophagy in tumor evasion from immune surveillance. Oncotarget 2016, 7, 17591–17607, doi:10.18632/oncotarget.7540.
- Zhu, H.; Wang, D.; Liu, Y.; Su, Z.; Zhang, L.; Chen, F.; Zhou, Y.; Wu, Y.; Yu, M.; Zhang, Z.; et al. Role of the Hypoxia-inducible factor-1 alpha induced autophagy in the conversion of non-stem pancreatic cancer cells into CD133+ pancreatic cancer stem-like cells. Cancer Cell Int. 2013, 13, 119, doi:10.1186/1475-2867-13-119.
- Cufi, S.; Vazquez-Martin, A.; Oliveras-Ferraros, C.; Martin-Castillo, B.; Vellon, L.; Menendez, J.A. Autophagy positively regulates the CD44(+) CD24(-/low) breast cancer stem-like phenotype. Cell Cycle 2011, 10, 3871–3885, doi:10.4161/cc.10.22.17976.
- Flynn, A.B.; Schiemann, W.P. Autophagy in breast cancer metastatic dormancy: Tumor suppressing or tumor promoting functions? J. Cancer Metastasis Treat. 2019, 5, doi:10.20517/2394-4722.2019.13.
- Guadamillas, M.C.; Cerezo, A.; Del Pozo, M.A. Overcoming anoikis--pathways to anchorage-independent growth in cancer. J. Cell Sci. 2011, 124, 3189–3197, doi:10.1242/jcs.072165.
- Gong, C.; Song, E.; Codogno, P.; Mehrpour, M. The roles of BECN1 and autophagy in cancer are context dependent. Autophagy 2012, 8, 1853–1855, doi:10.4161/auto.21996.
- Wolf, J.; Dewi, D.L.; Fredebohm, J.; Muller-Decker, K.; Flechtenmacher, C.; Hoheisel, J.D.; Boettcher, M. A mammosphere formation RNAi screen reveals that ATG4A promotes a breast cancer stem-like phenotype. Breast Cancer Res. 2013, 15, R109, doi:10.1186/bcr3576.
- Akalay, I.; Janji, B.; Hasmim, M.; Noman, M.Z.; Andre, F.; De Cremoux, P.; Bertheau, P.; Badoual, C.; Vielh, P.; Larsen, A.K.; et al. Epithelial-to-mesenchymal transition and autophagy induction in breast carcinoma promote escape from T-cell-mediated lysis. Cancer Res. 2013, 73, 2418–2427, doi:10.1158/0008-5472.CAN-12-2432.
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944, doi:10.1038/s41467-018-04070-6.
- Viry, E.; Baginska, J.; Berchem, G.; Noman, M.Z.; Medves, S.; Chouaib, S.; Janji, B. Autophagic degradation of GZMB/granzyme B: A new mechanism of hypoxic tumor cell escape from natural killer cell-mediated lysis. Autophagy 2014, 10, 173–175, doi:10.4161/auto.26924.
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455, doi:10.1073/pnas.1304790110.
- Petty, M.A.; Lo, E.H. Junctional complexes of the blood–brain barrier: Permeability changes in neuroinflammation. Progress in Neurobiology 2002, 68, 311–323, doi:10.1016/S0301-0082(02)00128-4.
- Armulik, A.; Genové, G.; Mäe, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561, doi:10.1038/nature09522.
- Horng, S.; Therattil, A.; Moyon, S.; Gordon, A.; Kim, K.; Argaw, A.T.; Hara, Y.; Mariani, J.N.; Sawai, S.; Flodby, P.; et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J. Clin. Invest. 2017, 127, 3136–3151, doi:10.1172/jci91301.
- Fazakas, C.; Wilhelm, I.; Nagyoszi, P.; Farkas, A.E.; Hasko, J.; Molnar, J.; Bauer, H.; Bauer, H.C.; Ayaydin, F.; Dung, N.T.; et al. Transmigration of melanoma cells through the blood-brain barrier: Role of endothelial tight junctions and melanoma-released serine proteases. PLoS ONE 2011, 6, e20758, doi:10.1371/journal.pone.0020758.
- Stamatovic, S.M.; Keep, R.F.; Andjelkovic, A.V. Brain endothelial cell-cell junctions: How to “open” the blood brain barrier. Curr. Neuropharmacol. 2008, 6, 179–192, doi:10.2174/157015908785777210.
- Bos, P.D.; Zhang, X.H.; Nadal, C.; Shu, W.; Gomis, R.R.; Nguyen, D.X.; Minn, A.J.; van de Vijver, M.J.; Gerald, W.L.; Foekens, J.A.; et al. Genes that mediate breast cancer metastasis to the brain. Nature 2009, 459, 1005–1009, doi:10.1038/nature08021.
- Miles, F.L.; Pruitt, F.L.; van Golen, K.L.; Cooper, C.R. Stepping out of the flow: Capillary extravasation in cancer metastasis. Clin. Exp. Metastasis 2008, 25, 305–324, doi:10.1007/s10585-007-9098-2.
- Mao, W.; Yi, X.; Qin, J.; Tian, M.; Jin, G. CXCL12/CXCR4 Axis Improves Migration of Neuroblasts Along Corpus Callosum by Stimulating MMP-2 Secretion After Traumatic Brain Injury in Rats. Neurochem. Res. 2016, 41, 1315–1322, doi:10.1007/s11064-016-1831-2.
- Virgintino, D.; Errede, M.; Rizzi, M.; Girolamo, F.; Strippoli, M.; Walchli, T.; Robertson, D.; Frei, K.; Roncali, L. The CXCL12/CXCR4/CXCR7 ligand-receptor system regulates neuro-glio-vascular interactions and vessel growth during human brain development. J. Inherit. Metab. Dis. 2013, 36, 455–466, doi:10.1007/s10545-012-9574-y.
- Terasaki, M.; Sugita, Y.; Arakawa, F.; Okada, Y.; Ohshima, K.; Shigemori, M. CXCL12/CXCR4 signaling in malignant brain tumors: A potential pharmacological therapeutic target. Brain Tumor Pathol. 2011, 28, 89–97, doi:10.1007/s10014-010-0013-1.
- Salmaggi, A.; Maderna, E.; Calatozzolo, C.; Gaviani, P.; Canazza, A.; Milanesi, I.; Silvani, A.; DiMeco, F.; Carbone, A.; Pollo, B. CXCL12, CXCR4 and CXCR7 expression in brain metastases. Cancer Biol. Ther. 2009, 8, 1608–1614, doi:10.4161/cbt.8.17.9202.
- Hinton, C.V.; Avraham, S.; Avraham, H.K. Role of the CXCR4/CXCL12 signaling axis in breast cancer metastasis to the brain. Clin. Exp. Metastasis 2010, 27, 97–105, doi:10.1007/s10585-008-9210-2.
- Holland, J.D.; Gyorffy, B.; Vogel, R.; Eckert, K.; Valenti, G.; Fang, L.; Lohneis, P.; Elezkurtaj, S.; Ziebold, U.; Birchmeier, W. Combined Wnt/beta-catenin, Met, and CXCL12/CXCR4 signals characterize basal breast cancer and predict disease outcome. Cell Rep. 2013, 5, 1214–1227, doi:10.1016/j.celrep.2013.11.001.
- Gil, M.; Seshadri, M.; Komorowski, M.P.; Abrams, S.I.; Kozbor, D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc. Natl. Acad. Sci. USA 2013, 110, E1291–1300, doi:10.1073/pnas.1220580110.
- Lobb, R.J.; Lima, L.G.; Moller, A. Exosomes: Key mediators of metastasis and pre-metastatic niche formation. Semin Cell Dev. Biol. 2017, 67, 3–10, doi:10.1016/j.semcdb.2017.01.004.
- Kienast, Y.; von Baumgarten, L.; Fuhrmann, M.; Klinkert, W.E.; Goldbrunner, R.; Herms, J.; Winkler, F. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010, 16, 116–122, doi:10.1038/nm.2072.
- Felding-Habermann, B.; O’’Toole, T.E.; Smith, J.W.; Fransvea, E.; Ruggeri, Z.M.; Ginsberg, M.H.; Hughes, P.E.; Pampori, N.; Shattil, S.J.; Saven, A.; et al. Integrin activation controls metastasis in human breast cancer. Proc. Natl. Acad. Sci. USA 2001, 98, 1853–1858, doi:10.1073/pnas.98.4.1853.
- Lorger, M.; Krueger, J.S.; O’Neal, M.; Staflin, K.; Felding-Habermann, B. Activation of tumor cell integrin alphavbeta3 controls angiogenesis and metastatic growth in the brain. Proc. Natl. Acad. Sci. USA 2009, 106, 10666–10671, doi:10.1073/pnas.0903035106.
- Avraham, H.K.; Jiang, S.; Fu, Y.; Nakshatri, H.; Ovadia, H.; Avraham, S. Angiopoietin-2 mediates blood-brain barrier impairment and colonization of triple-negative breast cancer cells in brain. J. Pathol. 2014, 232, 369–381, doi:10.1002/path.4304.
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341, doi:10.1016/j.ccell.2017.02.009.
- Heyn, C.; Ronald, J.A.; Ramadan, S.S.; Snir, J.A.; Barry, A.M.; MacKenzie, L.T.; Mikulis, D.J.; Palmieri, D.; Bronder, J.L.; Steeg, P.S.; et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med 2006, 56, 1001–1010, doi:10.1002/mrm.21029.
- Malladi, S.; Macalinao, D.G.; Jin, X.; He, L.; Basnet, H.; Zou, Y.; de Stanchina, E.; Massague, J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell 2016, 165, 45–60, doi:10.1016/j.cell.2016.02.025.
- Joosse, S.A.; Gorges, T.M.; Pantel, K. Biology, detection, and clinical implications of circulating tumor cells. EMBO Mol. Med. 2015, 7, 1–11, doi:10.15252/emmm.201303698.
- Giancotti, F.G. Mechanisms governing metastatic dormancy and reactivation. Cell 2013, 155, 750–764, doi:10.1016/j.cell.2013.10.029.
- De Angelis, M.L.; Francescangeli, F.; Zeuner, A. Breast Cancer Stem Cells as Drivers of Tumor Chemoresistance, Dormancy and Relapse: New Challenges and Therapeutic Opportunities. Cancers (Basel) 2019, 11, doi:10.3390/cancers11101569.
- Talukdar, S.; Bhoopathi, P.; Emdad, L.; Das, S.; Sarkar, D.; Fisher, P.B. Dormancy and cancer stem cells: An enigma for cancer therapeutic targeting. Adv. Cancer Res. 2019, 141, 43–84, doi:10.1016/bs.acr.2018.12.002.
- Gao, X.L.; Zhang, M.; Tang, Y.L.; Liang, X.H. Cancer cell dormancy: Mechanisms and implications of cancer recurrence and metastasis. Onco Targets Ther 2017, 10, 5219–5228, doi:10.2147/OTT.S140854.
- Shen, M.; Kang, Y. pSTAT3(+) Reactive Astrocytes Promote Brain Metastasis. Trends Mol. Med. 2018, 24, 733–735, doi:10.1016/j.molmed.2018.07.002.
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martinez, L.; Martinez-Saez, E.; Ramon, Y.C.S.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035, doi:10.1038/s41591-018-0044-4.
- Wasilewski, D.; Priego, N.; Fustero-Torre, C.; Valiente, M. Reactive Astrocytes in Brain Metastasis. Front. Oncol. 2017, 7, 298, doi:10.3389/fonc.2017.00298.
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405, doi:10.1038/nn.4493.
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487, doi:10.1038/nature21029.
- Jones, L.M.; Broz, M.L.; Ranger, J.J.; Ozcelik, J.; Ahn, R.; Zuo, D.; Ursini-Siegel, J.; Hallett, M.T.; Krummel, M.; Muller, W.J. STAT3 Establishes an Immunosuppressive Microenvironment during the Early Stages of Breast Carcinogenesis to Promote Tumor Growth and Metastasis. Cancer Res. 2016, 76, 1416–1428, doi:10.1158/0008-5472.CAN-15-2770.
- Moreira, M.P.; da Conceicao Braga, L.; Cassali, G.D.; Silva, L.M. STAT3 as a promising chemoresistance biomarker associated with the CD44(+/high)/CD24(-/low)/ALDH(+) BCSCs-like subset of the triple-negative breast cancer (TNBC) cell line. Exp. Cell Res. 2018, 363, 283–290, doi:10.1016/j.yexcr.2018.01.018.
- Blazquez, R.; Wlochowitz, D.; Wolff, A.; Seitz, S.; Wachter, A.; Perera-Bel, J.; Bleckmann, A.; Beissbarth, T.; Salinas, G.; Riemenschneider, M.J.; et al. PI3K: A master regulator of brain metastasis-promoting macrophages/microglia. Glia 2018, 66, 2438–2455, doi:10.1002/glia.23485.
- Solzak, J.P.; Atale, R.V.; Hancock, B.A.; Sinn, A.L.; Pollok, K.E.; Jones, D.R.; Radovich, M. Dual PI3K and Wnt pathway inhibition is a synergistic combination against triple negative breast cancer. NPJ Breast Cancer 2017, 3, 17, doi:10.1038/s41523-017-0016-8.
- Van Swearingen, A.E.D.; Sambade, M.J.; Siegel, M.B.; Sud, S.; McNeill, R.S.; Bevill, S.M.; Chen, X.; Bash, R.E.; Mounsey, L.; Golitz, B.T.; et al. Combined kinase inhibitors of MEK1/2 and either PI3K or PDGFR are efficacious in intracranial triple-negative breast cancer. Neuro Oncol. 2017, 19, 1481–1493, doi:10.1093/neuonc/nox052.
- Hohensee, I.; Lamszus, K.; Riethdorf, S.; Meyer-Staeckling, S.; Glatzel, M.; Matschke, J.; Witzel, I.; Westphal, M.; Brandt, B.; Muller, V.; et al. Frequent genetic alterations in EGFR- and HER2-driven pathways in breast cancer brain metastases. Am. J. Pathol. 2013, 183, 83–95, doi:10.1016/j.ajpath.2013.03.023.
- Zhang, L.; Zhang, S.; Yao, J.; Lowery, F.J.; Zhang, Q.; Huang, W.C.; Li, P.; Li, M.; Wang, X.; Zhang, C.; et al. Microenvironment-induced PTEN loss by exosomal microRNA primes brain metastasis outgrowth. Nature 2015, 527, 100–104, doi:10.1038/nature15376.
- Hohensee, I.; Chuang, H.N.; Grottke, A.; Werner, S.; Schulte, A.; Horn, S.; Lamszus, K.; Bartkowiak, K.; Witzel, I.; Westphal, M.; et al. PTEN mediates the cross talk between breast and glial cells in brain metastases leading to rapid disease progression. Oncotarget 2017, 8, 6155–6168, doi:10.18632/oncotarget.14047.
- Bahena-Ocampo, I.; Espinosa, M.; Ceballos-Cancino, G.; Lizarraga, F.; Campos-Arroyo, D.; Schwarz, A.; Maldonado, V.; Melendez-Zajgla, J.; Garcia-Lopez, P. miR-10b expression in breast cancer stem cells supports self-renewal through negative PTEN regulation and sustained AKT activation. EMBO Rep. 2016, 17, 648–658, doi:10.15252/embr.201540678.
- Conti, I.; Rollins, B.J. CCL2 (monocyte chemoattractant protein-1) and cancer. Semin Cancer Biol. 2004, 14, 149–154, doi:10.1016/j.semcancer.2003.10.009.
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and beta-Catenin and Akt activation. Mol. Cancer 2017, 16, 14, doi:10.1186/s12943-016-0570-y.
- Li, B.; Lu, Y.; Wang, H.; Han, X.; Mao, J.; Li, J.; Yu, L.; Wang, B.; Fan, S.; Yu, X.; et al. miR-221/222 enhance the tumorigenicity of human breast cancer stem cells via modulation of PTEN/Akt pathway. Biomed. Pharmacother. 2016, 79, 93–101, doi:10.1016/j.biopha.2016.01.045.
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E., 3rd; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163, doi:10.1073/pnas.0702596104.


