A higher propensity of developing brain metastasis exists in triple-negative breast cancer (TNBC). Upon comparing the metastatic patterns of all breast cancer subtypes, patients with TNBC exhibited increased risks of the brain being the initial metastatic site, early brain metastasis development, and shortest brain metastasis-related survival. Notably, the development of brain metastasis differs from that at other sites owing to the brain-unique microvasculature (blood brain barrier (BBB)) and intracerebral microenvironment. Studies of brain metastases from TNBC have revealed the poorest treatment response, mostly because of the relatively backward strategies to target vast disease heterogeneity and poor brain efficacy. Moreover, TNBC is highly associated with the existence of cancer stem cells (CSCs), which contribute to circulating cancer cell survival before BBB extravasation, evasion from immune surveillance, and plasticity in adaptation to the brain-specific microenvironment. We summarized recent literature regarding molecules and pathways and reviewed the effects of CSC biology during the formation of brain metastasis in TNBC.
- Cancer stem cell
- triple-negative breast cancer
- brain metastasis
- PTEN
- p-STAT3
- PI3K-Akt
- intracerebral niche
- blood brain barrier
1. Introduction
2. Triple-Negative Breast Cancer and Brain Metastasis
3. Understanding the Biology of Brain Metastases in Triple-Negative Breast Cancer
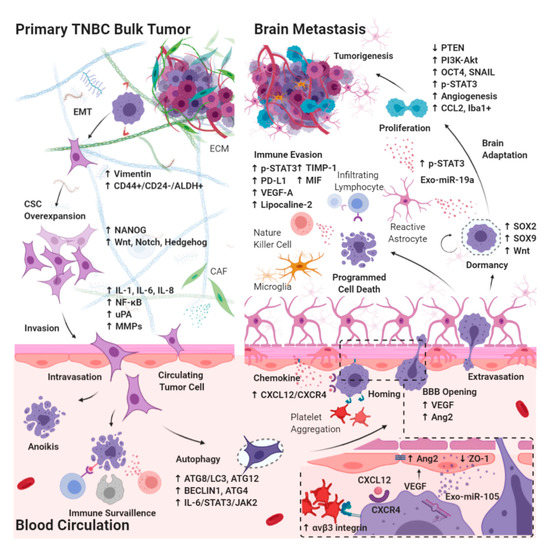
3.1. Effects of CSC Biology on BM-TNBC
3.1.1. Epithelial–Mesenchymal Transition in CSCs Breaking Away from the Primary Bulk Tumor
3.1.2. Autophagy in Circulating CSCs Undergoing Circulatory Arrest and Evading Immune Surveillance
3.2. Extravasation from the BBB
3.2.1. Chemokines Ligand Receptor System on Circulating CSCs Migrating through the BBB
3.2.2. Interaction between Brain Microvascular Endothelial Cells and Circulating CSCs Mediates the Tight Junction Disruption and BBB Destabilization
3.3. Intracerebral Tumor Microenvironment
3.3.1. Dormant Period of CSCs Adaptation to the Brain Microenvironment
3.3.2. Interaction between Reactive Astrocytes and CSCs Evading Immune Surveillance through the Activation of STAT3 Pathway
3.3.3. Tumor Progression through the Activation of PI3K/Akt Signaling by Interaction between Reactive Astrocytes and CSCs
3.3.4. Brain-Specific PTEN Suppression in Maintaining CSCs Plasticity during Tumorigenesis
This entry is adapted from the peer-reviewed paper 10.3390/cancers12082122