 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Dornatien Chuo Anang | + 2607 word(s) | 2607 | 2021-10-14 08:41:53 | | | |
| 2 | Lindsay Dong | + 172 word(s) | 2779 | 2021-10-27 05:06:59 | | |
Video Upload Options
Rheumatoid Arthritis (RA) is a chronic autoimmune disease characterized by joint inflammation, affecting approximately 1% of the general population. To alleviate symptoms and ameliorate joint damage, chronic use of immunosuppressives is needed. However, these treatments are only partially effective and may lead to unwanted side effects. Therefore, a more profound understanding of the pathophysiology might lead to more effective therapies, or better still, a cure. The presence of autoantibodies in RA indicates that B-cells might have a pivotal role in the disease. This concept is further supported by the fact that a diverse antibody response to various arthritis related epitopes is associated with arthritis development. In this context, attention has focused in recent years on the role of Germinal Centers (GCs) in RA. Since GCs act as the main anatomic location of somatic hypermutations, and thus contribute to the diversity and specificity of (auto) antibodies, it has been speculated that defects in germinal center reactions might be crucial in the initiation and maintenance of auto-immune events.
1. Introduction
2. Germinal Centers and Adaptive Immune Responses
GCs are specialized structures formed within LNs, spleen and other SLOs where affinity selection, clonal expansion and SHM of B cells takes place [15][16][17] (see Figure 1).
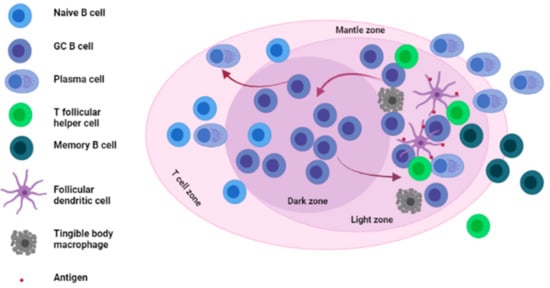
3. Germinal Centers in Rheumatoid Arthritis
There are several lines of evidence that implicate (ectopic) GCs in the process of autoantibody generation in RA. First and foremost are studies that have directly identified GCs in various tissues such as synovial tissues and lymph nodes of RA patients (discussed hereafter). A second line of evidence comes from examining RA-related autoantibodies. Multiple studies have demonstrated the presence of extensive SHM in these autoantibodies, which is a hallmark of a GC reaction. Thirdly, several groups have described defects in GC processes that result in features associated with auto-immunity.
3.1. The Presence of GCs and Ectopic GCs in Tissues of RA Patients and Mouse Models of Arthritis
3.1.1. The Presence of GCs and Ectopic GCs in Tissues of RA Patients
3.1.2. The Presence of GCs and Increased GC Responses in Tissues of RA Mouse Models
3.2. SHM within GCs and the Generation of RA-Related Autoantibodies
3.3. Defective GC-Resident Cellular Subsets and the Generation of Autoantibodies in RA
3.3.1. Defective Tfh Cell Functioning within GCs
| Tfh Cells in RA | Findings |
|---|---|
| Arroyo-Villa et al. [46] | RA patients with active disease have a higher frequency of Tfh cells and a higher Tfh/Tfr ratio resulting from lower Tfr frequencies |
| Zhang et al. [47] | Increased frequencies of Tfh cells and IL-21 in RA patients which correlates positively with DAS28 |
| Wang et al. [48], Zhou et al. [50] |
Increased frequencies of Tfh cells in newly diagnosed RA patients correlating with activated B cells and DAS28 |
| Niu et al. [49] | Elevated frequencies of Tfh cells, IL-21 and PD-1 in RA patients with active disease |
| Nakayamada et al. [51] | Higher proportions of Tfh cells in RA patients with active disease and, treatment with Abatacept reduces Tfh cell levels. |
| Su et al. [52] | Elevated frequencies of Tfh cells in RA patients compared to HCs |
| Tfr cells in RA | |
| Su et al. [52] | Decreased frequencies of Tfr cells in RA patients compared to HCs |
| Wang et al. [53] | Increased levels of Tfr cells in RA patients compared to HCs |
| Macdonald et al. [54] | Elevated percentages of Tfr cells in RA patients |
| Liu et al. [55] | Higher frequencies of Tfr and Tfr/Tfh cell ratio in RA patients with stable remission |
3.3.2. Defective Tfr Cell Functioning within GCs
References
- Silman, A.J.; Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res. 2002, 4, S265–S272.
- Shapira, Y.; Agmon-Levin, N.; Shoenfeld, Y. Geoepidemiology of autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2010, 6, 468–476.
- Edwards, J.C.; Szczepanski, L.; Szechinski, J.; Filipowicz-Sosnowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2581.
- Gerlag, D.M.; Safy, M.; Maijer, K.I.; Tang, M.W.; Tas, S.W.; Starmans-Kool, M.J.F.; van Tubergen, A.; Janssen, M.; de Hair, M.; Hansson, M.; et al. Effects of B-cell directed therapy on the preclinical stage of rheumatoid arthritis: The PRAIRI study. Ann. Rheum. Dis. 2019, 78, 179–185.
- Van der Woude, D.; Rantapaa-Dahlqvist, S.; Ioan-Facsinay, A.; Onnekink, C.; Schwarte, C.M.; Verpoort, K.N.; Drijfhout, J.W.; Huizinga, T.W.; Toes, R.E.; Pruijn, G.J. Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann. Rheum. Dis. 2010, 69, 1554–1561.
- Van de Stadt, L.A.; van der Horst, A.R.; de Koning, M.H.; Bos, W.H.; Wolbink, G.J.; van de Stadt, R.J.; Pruijn, G.J.; Dijkmans, B.A.; van Schaardenburg, D.; Hamann, D. The extent of the anti-citrullinated protein antibody repertoire is associated with arthritis development in patients with seropositive arthralgia. Ann. Rheum. Dis. 2011, 70, 128–133.
- Hozumi, N.; Tonegawa, S. Evidence for somatic rearrangement of immunoglobulin genes coding for variable and constant regions. Proc. Natl. Acad. Sci. USA 1976, 73, 3628–3632.
- Vinuesa, C.G.; Sanz, I.; Cook, M.C. Dysregulation of germinal centres in autoimmune disease. Nat. Rev. Immunol. 2009, 9, 845–857.
- Kim, H.-J.; Krenn, V.; Steinhauser, G.; Berek, C. Plasma Cell Development in Synovial Germinal Centers in Patients with Rheumatoid and Reactive Arthritis. J. Immunol. 1999, 162, 3053–3062.
- Wagner, U.G.; Kurtin, P.J.; Wahner, A.; Brackertz, M.; Berry, D.J.; Goronzy, J.J.; Weyand, C.M. The role of CD8+ CD40L+ T cells in the formation of germinal centers in rheumatoid synovitis. J. Immunol. 1998, 161, 6390–6397.
- Luzina, I.G.; Atamas, S.P.; Storrer, C.E.; daSilva, L.C.; Kelsoe, G.; Papadimitriou, J.C.; Handwerger, B.S. Spontaneous formation of germinal centers in autoimmune mice. J. Leukoc. Biol. 2001, 70, 578–584.
- Domeier, P.P.; Schell, S.L.; Rahman, Z.S.M. Spontaneous germinal centers and autoimmunity. Autoimmunity 2017, 50, 4–18.
- Mietzner, B.; Tsuiji, M.; Scheid, J.; Velinzon, K.; Tiller, T.; Abraham, K.; Gonzalez, J.B.; Pascual, V.; Stichweh, D.; Wardemann, H.; et al. Autoreactive IgG memory antibodies in patients with systemic lupus erythematosus arise from nonreactive and polyreactive precursors. Proc. Natl. Acad. Sci. USA 2008, 105, 9727–9732.
- Di Zenzo, G.; Di Lullo, G.; Corti, D.; Calabresi, V.; Sinistro, A.; Vanzetta, F.; Didona, B.; Cianchini, G.; Hertl, M.; Eming, R.; et al. Pemphigus autoantibodies generated through somatic mutations target the desmoglein-3 cis-interface. J. Clin. Investig. 2012, 122, 3781–3790.
- Allen, C.D.C.; Cyster, J.G. Follicular dendritic cell networks of primary follicles and germinal centers: Phenotype and function. Semin. Immunol. 2008, 20, 14–25.
- Berek, C.; Berger, A.; Apel, M. Maturation of the immune response in germinal centers. Cell 1991, 67, 1121–1129.
- Arulraj, T.; Binder, S.C.; Robert, P.A.; Meyer-Hermann, M. Germinal Centre Shutdown. Front. Immunol. 2021, 12, 2730.
- Bombardieri, M.; Lewis, M.; Pitzalis, C. Ectopic lymphoid neogenesis in rheumatic autoimmune diseases. Nat. Rev. Rheumatol. 2017, 13, 141–154.
- Humby, F.; Bombardieri, M.; Manzo, A.; Kelly, S.; Blades, M.C.; Kirkham, B.; Spencer, J.; Pitzalis, C. Ectopic lymphoid structures support ongoing production of class-switched autoantibodies in rheumatoid synovium. PLoS Med. 2009, 6, e1.
- Takemura, S.; Braun, A.; Crowson, C.; Kurtin, P.J.; Cofield, R.H.; O’Fallon, W.M.; Goronzy, J.J.; Weyand, C.M. Lymphoid neogenesis in rheumatoid synovitis. J. Immunol. 2001, 167, 1072–1080.
- Shipman, W.D.; Dasoveanu, D.C.; Lu, T.T. Tertiary lymphoid organs in systemic autoimmune diseases: Pathogenic or protective? F1000Research 2017, 6, 196.
- Mellors, R.C.; Heimer, R.; Corcos, J.; Korngold, L. Cellular origin of rheumatoid factor. J. Exp. Med. 1959, 110, 875–886.
- Imai, Y.; Sato, T.; Yamakawa, M.; Kasajima, T.; Suda, A.; Watanabe, Y. A morphological and immunohistochemical study of lymphoid germinal centers in synovial and lymph node tissues from rheumatoid arthritis patients with special reference to complement components and their receptors. Acta Pathol. Jpn. 1989, 39, 127–134.
- Randen, I.; Thompson, K.M.; Natvig, J.B.; Førre, O.; Waalen, K. Human monoclonal rheumatoid factors derived from the polyclonal repertoire of rheumatoid synovial tissue: Production and characterization. Clin. Exp. Immunol. 1989, 78, 13–18.
- Dahdah, A.; Habir, K.; Nandakumar, K.S.; Saxena, A.; Xu, B.; Holmdahl, R.; Malin, S. Germinal Center B Cells Are Essential for Collagen-Induced Arthritis. Arthritis Rheumatol. 2018, 70, 193–203.
- Louis, C.; Ngo, D.; D’Silva, D.B.; Hansen, J.; Phillipson, L.; Jousset, H.; Novello, P.; Segal, D.; Lawlor, K.E.; Burns, C.J.; et al. Therapeutic Effects of a TANK-Binding Kinase 1 Inhibitor in Germinal Center-Driven Collagen-Induced Arthritis. Arthritis Rheumatol. 2019, 71, 50–62.
- Han, S.; Cao, S.; Bheekha-Escura, R.; Zheng, B. Germinal center reaction in the joints of mice with collagen-induced arthritis: An animal model of lymphocyte activation and differentiation in arthritis joints. Arthritis Rheum. 2001, 44, 1438–1443.
- Hou, L.; Block, K.E.; Huang, H. Artesunate abolishes germinal center B cells and inhibits autoimmune arthritis. PLoS ONE 2014, 9, e104762.
- Wang, J.H.; New, J.S.; Xie, S.; Yang, P.; Wu, Q.; Li, J.; Luo, B.; Ding, Y.; Druey, K.M.; Hsu, H.-C.; et al. Extension of the germinal center stage of B cell development promotes autoantibodies in BXD2 mice. Arthritis Rheum. 2013, 65, 2703–2712.
- Guo, W.; Smith, D.; Aviszus, K.; Detanico, T.; Heiser, R.A.; Wysocki, L.J. Somatic hypermutation as a generator of antinuclear antibodies in a murine model of systemic autoimmunity. J. Exp. Med. 2010, 207, 2225–2237.
- Ray, S.K.; Putterman, C.; Diamond, B. Pathogenic autoantibodies are routinely generated during the response to foreign antigen: A paradigm for autoimmune disease. Proc. Natl. Acad. Sci. USA 1996, 93, 2019–2024.
- Li, S.; Yu, Y.; Yue, Y.; Liao, H.; Xie, W.; Thai, J.; Mikuls, T.R.; Thiele, G.M.; Duryee, M.J.; Sayles, H.; et al. Autoantibodies from Single Circulating Plasmablasts React with Citrullinated Antigens and Porphyromonas gingivalis in Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 614–626.
- Xu, X.; Hsu, H.C.; Chen, J.; Grizzle, W.E.; Chatham, W.W.; Stockard, C.R.; Wu, Q.; Yang, P.A.; Holers, V.M.; Mountz, J.D. Increased expression of activation-induced cytidine deaminase is associated with anti-CCP and rheumatoid factor in rheumatoid arthritis. Scand. J. Immunol. 2009, 70, 309–316.
- Gatto, D.; Brink, R. The germinal center reaction. J. Allergy Clin. Immunol. 2010, 126, 898–907.
- Bugatti, S.; Manzo, A.; Vitolo, B.; Fusetti, C.; Humby, F.; Caporali, R.; Pitzalis, C.; Montecucco, C. B cell distribution and activation-induced cytidine deaminase expression in rheumatoid synovitis: Clinical and bio-molecular correlates. J. Ann. Rheum. Dis. 2011, 70, A55.
- Sabouri, Z.; Schofield, P.; Horikawa, K.; Spierings, E.; Kipling, D.; Randall, K.L.; Langley, D.; Roome, B.; Vazquez-Lombardi, R.; Rouet, R.; et al. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc. Natl. Acad. Sci. USA 2014, 111, E2567–E2575.
- Reed, J.H.; Jackson, J.; Christ, D.; Goodnow, C.C. Clonal redemption of autoantibodies by somatic hypermutation away from self-reactivity during human immunization. J. Exp. Med. 2016, 213, 1255–1265.
- Weber, J.P.; Fuhrmann, F.; Feist, R.K.; Lahmann, A.; Al Baz, M.S.; Gentz, L.-J.; Vu Van, D.; Mages, H.W.; Haftmann, C.; Riedel, R.; et al. ICOS maintains the T follicular helper cell phenotype by down-regulating Krüppel-like factor 2. J. Exp. Med. 2015, 212, 217–233.
- Meyer-Hermann, M.E.; Maini, P.K.; Iber, D. An analysis of B cell selection mechanisms in germinal centers. Math. Med. Biol. A J. IMA 2006, 23, 255–277.
- Cao, G.; Chi, S.; Wang, X.; Sun, J.; Zhang, Y. CD4+CXCR5+PD-1+ T Follicular Helper Cells Play a Pivotal Role in the Development of Rheumatoid Arthritis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2019, 25, 3032–3040.
- Chevalier, N.; Macia, L.; Tan, J.K.; Mason, L.J.; Robert, R.; Thorburn, A.N.; Wong, C.H.; Tsai, L.M.; Bourne, K.; Brink, R.; et al. The Role of Follicular Helper T Cell Molecules and Environmental Influences in Autoantibody Production and Progression to Inflammatory Arthritis in Mice. Arthritis Rheumatol. 2016, 68, 1026–1038.
- Moschovakis, G.L.; Bubke, A.; Friedrichsen, M.; Falk, C.S.; Feederle, R.; Forster, R. T cell specific Cxcr5 deficiency prevents rheumatoid arthritis. Sci. Rep. 2017, 7, 8933.
- Jang, E.; Cho, S.H.; Park, H.; Paik, D.J.; Kim, J.M.; Youn, J. A positive feedback loop of IL-21 signaling provoked by homeostatic CD4+CD25- T cell expansion is essential for the development of arthritis in autoimmune K/BxN mice. J. Immunol. 2009, 182, 4649–4656.
- Morita, R.; Schmitt, N.; Bentebibel, S.-E.; Ranganathan, R.; Bourdery, L.; Zurawski, G.; Foucat, E.; Dullaers, M.; Oh, S.; Sabzghabaei, N.; et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity 2011, 34, 108–121.
- Crotty, S. Follicular helper CD4 T cells (TFH). Annu. Rev. Immunol. 2011, 29, 621–663.
- Arroyo-Villa, I.; Bautista-Caro, M.-B.; Balsa, A.; Aguado-Acín, P.; Bonilla-Hernán, M.-G.; Plasencia, C.; Villalba, A.; Nuño, L.; Puig-Kröger, A.; Martín-Mola, E.; et al. Constitutively altered frequencies of circulating follicullar helper T cell counterparts and their subsets in rheumatoid arthritis. Arthritis Res. Ther. 2014, 16, 500.
- Zhang, N.; Zhao, P.; Shrestha, A.; Zhang, L.; Qu, Z.; Liu, M.; Zhang, S.; Jiang, Y. A higher frequency of CD4+CXCR5+ T follicular helper cells in adult patients with minimal change disease. BioMed. Res. Int. 2014, 2014, 836157.
- Wang, J.; Shan, Y.; Jiang, Z.; Feng, J.; Li, C.; Ma, L.; Jiang, Y. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Exp. Immunol. 2013, 174, 212–220.
- Niu, Q.; Huang, Z.C.; Wu, X.J.; Jin, Y.X.; An, Y.F.; Li, Y.M.; Xu, H.; Yang, B.; Wang, L.L. Enhanced IL-6/phosphorylated STAT3 signaling is related to the imbalance of circulating T follicular helper/T follicular regulatory cells in patients with rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 200.
- Zhou, H.; Hu, B.; Zhaopeng, Z.; Liu, J.; Zhong, Q.; Fan, Y.; Li, L. Elevated circulating T cell subsets and cytokines expression in patients with rheumatoid arthritis. Clin. Rheumatol. 2019, 38, 1831–1839.
- Nakayamada, S.; Kubo, S.; Yoshikawa, M.; Miyazaki, Y.; Yunoue, N.; Iwata, S.; Miyagawa, I.; Hirata, S.; Nakano, K.; Saito, K.; et al. Differential effects of biological DMARDs on peripheral immune cell phenotypes in patients with rheumatoid arthritis. Rheumatology 2018, 57, 164–174.
- Su, R.; Wang, Y.; Hu, F.; Li, B.; Guo, Q.; Zheng, X.; Liu, Y.; Gao, C.; Li, X.; Wang, C. Altered Distribution of Circulating T Follicular Helper-Like Cell Subsets in Rheumatoid Arthritis Patients. Front. Med. 2021, 8, 1024.
- Wang, X.; Yang, C.; Xu, F.; Qi, L.; Wang, J.; Yang, P. Imbalance of circulating Tfr/Tfh ratio in patients with rheumatoid arthritis. Clin. Exp. Med. 2019, 19, 55–64.
- Macdonald, A.J.; Cerosaletti, K.; Chen, J.; Nguyen, T.-S.; Posso, S.; Marchesini, G.; Abashian, M.; Martin, A.P.; De Rosa, D.; Samanta, T.; et al. OP0264 Relative Frequencies of Circulating T Follicular Helper and T Follicular Regulatory Cells in Autoimmune Patients and Healthy Control Donors and The Effect of Disease Modulating Therapy. J. Ann. Rheum. Dis. 2016, 75, 158.
- Liu, C.; Wang, D.; Lu, S.; Xu, Q.; Zhao, L.; Zhao, J.; Song, Y.; Wang, H. Increased Circulating Follicular Treg Cells Are Associated with Lower Levels of Autoantibodies in Patients with Rheumatoid Arthritis in Stable Remission. Arthritis Rheumatol. 2018, 70, 711–721.
- Sage, P.T.; Sharpe, A.H. T follicular regulatory cells in the regulation of B cell responses. Trends Immunol. 2015, 36, 410–418.
- Gong, Y.; Tong, J.; Wang, S. Are Follicular Regulatory T Cells Involved in Autoimmune Diseases? Front. Immunol. 2017, 8, 1790.
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982.
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988.
- Fonseca, V.R.; Agua-Doce, A.; Maceiras, A.R.; Pierson, W.; Ribeiro, F.; Romao, V.C.; Pires, A.R.; da Silva, S.L.; Fonseca, J.E.; Sousa, A.E.; et al. Human blood Tfr cells are indicators of ongoing humoral activity not fully licensed with suppressive function. Sci. Immunol. 2017, 2.
- Zhu, Y.; Zou, L.; Liu, Y.-C. T follicular helper cells, T follicular regulatory cells and autoimmunity. Int. Immunol. 2016, 28, 173–179.
- Laidlaw, B.J.; Lu, Y.; Amezquita, R.A.; Weinstein, J.S.; Vander Heiden, J.A.; Gupta, N.T.; Kleinstein, S.H.; Kaech, S.M.; Craft, J. Interleukin-10 from CD4(+) follicular regulatory T cells promotes the germinal center response. Sci. Immunol. 2017, 2.
- Ding, Y.; Li, J.; Yang, P.; Luo, B.; Wu, Q.; Zajac, A.J.; Wildner, O.; Hsu, H.-C.; Mountz, J.D. Interleukin-21 Promotes Germinal Center Reaction by Skewing the Follicular Regulatory T Cell to Follicular Helper T Cell Balance in Autoimmune BXD2 Mice. Arthritis Rheumatol. 2014, 66, 2601–2612.
- Fonseca, V.R.; Romao, V.C.; Agua-Doce, A.; Santos, M.; Lopez-Presa, D.; Ferreira, A.C.; Fonseca, J.E.; Graca, L. The Ratio of Blood T Follicular Regulatory Cells to T Follicular Helper Cells Marks Ectopic Lymphoid Structure Formation While Activated Follicular Helper T Cells Indicate Disease Activity in Primary Sjogren’s Syndrome. Arthritis Rheumatol. 2018, 70, 774–784.
- Shan, Y.; Qi, C.; Zhao, J.; Liu, Y.; Gao, H.; Zhao, D.; Ding, F.; Wang, J.; Jiang, Y. Higher frequency of peripheral blood follicular regulatory T cells in patients with new onset ankylosing spondylitis. Clin. Exp. Pharmacol. Physiol. 2015, 42, 154–161.
- Fu, W.; Liu, X.; Lin, X.; Feng, H.; Sun, L.; Li, S.; Chen, H.; Tang, H.; Lu, L.; Jin, W.; et al. Deficiency in T follicular regulatory cells promotes autoimmunity. J. Exp. Med. 2018, 215, 815–825.
- Miyabe, C.; Miyabe, Y.; Strle, K.; Kim, N.D.; Stone, J.H.; Luster, A.D.; Unizony, S. An expanded population of pathogenic regulatory T cells in giant cell arteritis is abrogated by IL-6 blockade therapy. Ann. Rheum. Dis. 2017, 76, 898–905.



