Rheumatoid Arthritis (RA) is a chronic autoimmune disease characterized by joint inflammation, affecting approximately 1% of the general population. To alleviate symptoms and ameliorate joint damage, chronic use of immunosuppressives is needed. However, these treatments are only partially effective and may lead to unwanted side effects. Therefore, a more profound understanding of the pathophysiology might lead to more effective therapies, or better still, a cure.
The presence of autoantibodies in RA indicates that B-cells might have a pivotal role in the disease. This concept is further supported by the fact that a diverse antibody response to various arthritis related epitopes is associated with arthritis development. In this context, attention has focused in recent years on the role of Germinal Centers (GCs) in RA. Since GCs act as the main anatomic location of somatic hypermutations, and thus contribute to the diversity and specificity of (auto) antibodies, it has been speculated that defects in germinal center reactions might be crucial in the initiation and maintenance of auto-immune events. In this paper, we discuss current evidence that various processes within GCs can result in the aberrant production of B cells that possess autoreactive properties and might result in the production of RA related autoantibodies. Secondly, we discuss various (pre-)clinical studies that have targeted various GC processes as novel therapies for RA treatment.
1. Introduction
RA is a common autoimmune disease that affects about 1% of the worlds’ population [
1,
2]. Despite the fact that the disease pathogenesis is poorly understood, the clinical efficacy of B cell-directed therapy (BCDT) as well as findings that B cell depletion can delay onset of the disease underscore the concept that B cells are likely to play a pivotal role in the disease process [
3,
4]. Additionally, the finding that epitope spreading of the antibody response is associated with disease onset and disease course in early arthritis suggest that a more diverse antibody and B cell immune response is crucial for disease development [
5,
6]. This hypothesis suggests that the key to understanding the role of B cells in RA lies at an earlier stage of B cell development and differentiation. The question of when and where these B cells of varied specificities arise has largely remained unanswered for decades. Interestingly, somatic hyper mutation (SHM) of V(D)J genes, which is known to be the major contributor to Immunoglobulin (Ig) diversity and specificity, occurs primarily within GCs [
7]. Whether the process of SHM and other processes within GCs are the main drivers of autoimmunity is yet to be fully explored. Therefore, there is an urgent need to unravel in detail the GC responses, as this might be the origin of not only anti-citrullinated protein antibodies (ACPA) producing B cells but also other B cells of unknown specificities that largely contribute to the chronic inflammatory process in RA as well as other autoimmune diseases.
More recently, compelling evidence has pointed to GCs as a possible source of these autoreactive B cells in RA and other autoimmune disorders [
17,
18,
19,
20,
21]. More so, the findings that reverting most autoantibodies to their germline configuration results in loss of their reactivity towards their target antigens as well as recent advances in high throughput technologies that permit a high-resolution dissection of GCs and its components has spurred enormous interest into GC biology as a potential culprit in the generation of these autoantibodies [
22,
23]. Therefore, a deeper understanding of GC responses in the context of autoimmunity is needed and could be a next step towards modulating those immune changes that are a major signature of these diseases.
2. Germinal Centers and Adaptive Immune Responses
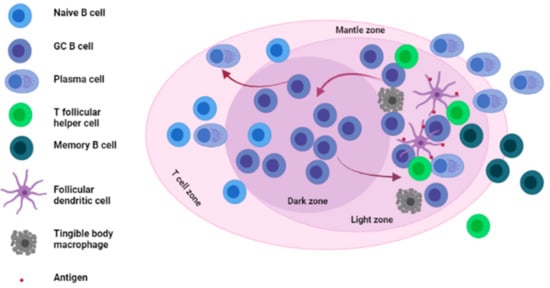
GCs are specialized structures formed within LNs, spleen and other SLOs where affinity selection, clonal expansion and SHM of B cells takes place [24,25,26] (see Figure 1).
Figure 1. Architecture and main cellular components of the germinal center. The germinal center is mainly divided into two zones; the Dark zone (DZ) where GC B cells undergo somatic hypermutation (SHM) of their immunoglobulin (Ig) genes and the light zone (LZ) where GC B cells interact with antigens presented to them by FDCs as well as T follicular helper cells (Tfh). GC: germinal center, FDCs: follicular dendritic cells.
3. Germinal Centers in Rheumatoid Arthritis
There are several lines of evidence that implicate (ectopic) GCs in the process of autoantibody generation in RA. First and foremost are studies that have directly identified GCs in various tissues such as synovial tissues and lymph nodes of RA patients (discussed hereafter). A second line of evidence comes from examining RA-related autoantibodies. Multiple studies have demonstrated the presence of extensive SHM in these autoantibodies, which is a hallmark of a GC reaction. Thirdly, several groups have described defects in GC processes that result in features associated with auto-immunity.
3.1. The Presence of GCs and Ectopic GCs in Tissues of RA Patients and Mouse Models of Arthritis
3.1.1. The Presence of GCs and Ectopic GCs in Tissues of RA Patients
While traditional GCs have gained enormous interest in recent years, ectopic GCs have been reported in about 40–60% of RA patients as well [
38,
39]. These ectopic GCs, which form primarily at sites of inflammation, are known to differ from traditional GCs in their vasculature, microenvironment and composition. In the literature, these ectopic GCs are thought to intensify or continue the autoimmune response outside the follicular region or arise as a feedback mechanism aimed at censoring the autoreactive B cell clones that arise as by-products from traditional GC reactions [
40,
41]. Both GCs and ectopic GCs have been implicated in the pathogenesis of RA. The findings that rheumatoid factors (RFs) could be found within GCs in lymph nodes obtained from RA patients more than fifty years ago was one of the first pieces of evidence of a possible involvement of GCs in the development of RA [
42]. Another indication of GC involvement in RA was reported in the late 1980s. Using microscopy and immunohistochemistry, Imai et al. reported increased numbers of GCs in lymph nodes and synovial tissues of RA patients [
43]. In recent years, evidence has come from observations in synovial tissues and lymph nodes of RA patients [
18,
44]. In one of such studies, microdissection and sequencing of V
H/V
L chains of synovial tissue ectopic GC-derived B cells revealed a high degree of clonally related plasma cells, suggesting the possibility that these cells underwent terminal differentiation in the inflamed synovium of this patients. These results indicate that local GCs as well as ectopic GCs may, indeed, contribute to the pool of antibody-producing plasma cells that accumulate at a local site of inflammation.
3.1.2. The Presence of GCs and Increased GC Responses in Tissues of RA Mouse Models
In addition to observations in human RA, findings from animal models of arthritis have reported clues on the role of GCs in animal models of RA. In the well-characterized collagen-induced arthritis (CIA) mouse model of arthritis, the development of autoimmune arthritis was highly associated with an increased GC response in lymph node tissues [
45]. Interestingly, an increased GC response in these mice correlated with an increased number of GC-B cells as well as an increased production of anti-collagen antibodies. In another study involving an antigen-induced arthritis (AIA) mouse model, a K/BxN serum-transfer–induced arthritis (STIA) mouse model, and CIA mice, an increased traditional GC response was observed in all mouse models compared to wild type controls and, inhibiting GC reactions resulted in a marked decrease in antibody-driven arthritis [
46]. Finally, several other studies from Han et al., Hou et al., and Wang et al. have all reported similar results implicating GCs and GC reactions in the disease process in animal models of RA [
47,
48,
49].
3.2. SHM within GCs and the Generation of RA-Related Autoantibodies
Another finding that implicates GCs in RA development is the presence of SHM in RA-related autoantibodies. SHM, a process known to occur on centroblasts within GCs often results in the accumulation of point mutations in the Ig variable genes of antibodies. While SHM often results in the production of B cell clones with high affinity B cell receptors (BcRs), the process of SHM is not able to distinguish between “favorable” and “unfavorable” mutations. It is the resulting BcRs affinity that is the basis for affinity selection. Therefore, B cells with varied BcR affinities and specificities result from this process.
As the SHM process is stochastic in nature, a possible outcome could be the ‘accidental’ generation and positive selection of B cell clones with high affinity for self-antigens [
50,
51]. The subsequent exit of such high affinity self-reactive B cell clones from the GC into the periphery could result in the initiation of autoimmune events. A compelling example outlining this possibility was a study by Guo et al., which reported the creation of high-avidity autoimmune B cells from non-autoimmune precursors by the process of SHM targeted in V region genes of anti-nuclear antibody (ANA) producing B cells [
50]. In the context of RA, a strong correlation between SHM and the generation of ACPAs came from a report by Li et al., where the presence of highly somatically mutated Ig genes encoding ACPAs in RA patients was reported. More interesting, reverting the variable regions of these ACPA encoding Ig genes to their respective germline genes resulted in loss of their reactivity towards citrullinated RA antigens further indicating a possible GC origin of these antibodies [
52].
While the studies discussed above directly implicate the process of SHM in autoantibody generation, there are additional findings on Activation-induced Cytidine Deaminase (AID), which is a key enzyme in regulating SHM that implicates SHM in autoimmunity. In a group of RA patients, an increased expression of AID was reported in synovial tissues and peripheral blood [
53]. Additionally, a correlation was observed between AID expression and the production of IgG and IgM RF and other autoantibodies against citrullinated peptides. Two other studies have also described the presence of cells which carrySHMs in fibroblast-like synoviocytes (FLS) and synovium of RA individuals [
32,
54]. Additionally in a study involving BXD2 mice, a mouse model for autoimmune lupus, AID was shown to be highly expressed in splenic tissues and the upregulation could be correlated with the generation of pathogenic autoantibodies in GCs in these mice [
49].
Finally, it is important to note that while the process of SHM can largely contribute to the generation of autoreactive B cells, recent evidence indicates that point mutations introduced by SHM on the other hand can circumvent autoantibody production by redeeming self-reactive B cells recruited into GCs [
55,
56]. Further investigation is needed to explore this possibility.
3.3. Defective GC-Resident Cellular Subsets and the Generation of Autoantibodies in RA
Although GCs are difficult to study as they are multicellular structures, there is evidence that defects in GCs are associated with auto-immunity. Here, we will discuss the available observations on Tfh and Tfr cells as these GCs cells have received much interest recently in the context of RA and seem likely candidates for targeted therapeutic strategies.
3.3.1. Defective Tfh Cell Functioning within GCs
Within GCs, Tfh cells are known to be crucial players in the process of affinity selection, which takes place in the LZ. This function is mediated through ICOS (a CD28 family receptor expressed on their surface), CD40L and through their signature cytokines IL-21 and IL-4 [
57]. Limiting the number of Tfh cells within a GC reaction was observed to be crucial in the selection of B cells with high affinity BcRs [
58]. Therefore, it seems that Tfh quantity and quality are a major limiting factor for B cell selection within GCs. Hence, aberrant or excessive Tfh signals within GCs could result in a GC environment characterized by reduced competition from B cells. The outcome is the selection of B cells carrying both “normal” as well as “autoreactive” BcRs.
There is evidence of such aberrant Tfh numbers in various RA mouse models. Recently, Cao and colleagues reported an increased frequency of Tfh cells in CIA mice compared to unimmunized animals. Furthermore, this increase was associated with elevated levels of (interleukin-21) IL-21, a signature cytokine produced by Tfh cells [
59]. Independently, two groups demonstrated the role of Tfh cell-associated markers (CXCR5 and adaptor SLAM-associated protein (SAP)) in the induction and maintenance of arthritis in CIA as well as SAP
−/− KRN-Tg mice models. In both studies, arthritis was prevented when these molecules were targeted [
60,
61]. In addition, another study in K/BxN mouse described the importance of IL-21, a typical Tfh cytokine in the development of arthritis. In this study, an induced deficiency in the receptor of IL-21 was sufficient to completely prevent the onset and subsequent development of arthritis in the K/BxN mouse model. This clearly attributes an important role of the IL-21/IL-21R axis in the very early stages of arthritis development [
62].
It is worth mentioning that unlike in mouse models, difficulties do exist in assessing GC-Tfh cells in humans due to their relatively low numbers coupled with obstacles in tissue sampling. Therefore, the circulatory form of these cells termed “Circulatory T follicular helper cells (cTfh)” have been extensively described as a counterpart of Tfh cells in GCs [
63,
64]. Data from human studies on cTfh cells in RA patients clearly corroborate mouse data on aberrant Tfh numbers (
Table 1). Several studies have reported an increase in cTfh cells in RA patients, accompanied by elevated IL-21 levels in some cases [
65,
66,
67,
68,
69,
70,
71]. However, it is important to note that while there are increase Tfh numbers both in murine and human GCs, the underlying mechanism responsible for their expansion remains to be fully unraveled. Could antigen quantity and quality within GCs be responsible for their expansion? Or do these cells expand already in the periphery prior to their migration into GCs? These are some of the questions that will need to be answered.
3.3.2. Defective Tfr Cell Functioning within GCs
In recent years, a regulatory GC-resident T cell subset termed T follicular regulatory cells (Tfr) has been in the spotlight. Similar to Tfh cells, Tfr cells express CXCR5, Bcl-6, ICOS and PD-1 coupled with their expression of Foxp3 and CTLA-4, which enables them to be distinguished from Tfh cells [
75,
76]. Since their initial discovery more than a decade ago, a major point of contention has been the development and maturation process of Tfr cells. While an earlier study from Linterman and colleagues suggested that thymic Tregs have the ability to convert into Tfr cells [
77], this was disputed by two other studies which reported the absence of Tfr cells in the thymus [
78,
79].
Tfr cells have generally been attributed with regulating immune reactions in the GC by dampening excessive GC responses through their interactions with Tfh cells [
80]. On the other side, there is evidence of their ability to enhance GC reactions by helping B cells through the release of IL-10 [
81]. The mechanism by which Tfr cells can execute both functions of suppressing autoimmunity and promoting B cell help simultaneously have largely remained unexplored. Nevertheless, accumulated evidence has demonstrated that abnormal Tfr cells may result in an imbalance immune milieu that favors the development of autoimmune diseases [
79,
82,
83,
84,
85]. Two studies exploring the contribution of Tfr cells in RA reported similar findings on elevated Tfr levels in RA patients compared to healthy controls [
72,
73]. In addition, the regulatory function of Tfr cells was revealed in a recent study by Liu et al., who found an association of increased Tfr cells with decreased autoantibody production in RA patients in stable remission [
74].
Finally, while their name suggests a cell type involved in immune regulation, it is not clear as to whether the reported elevated levels of Tfr in RA patients is a feedback mechanism aimed at dampening potential “abnormal” Tfh responses in the GCs or on their own contribute to the disease pathogenesis. The rationale of the last option is supported by the observation that a regulatory cell type on its own could be potential pathogenic in another milieu, as has been reported in giant cell arteritis [
86].
This entry is adapted from the peer-reviewed paper 10.3390/ijms221910514