Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kanako Miyano | + 2310 word(s) | 2310 | 2021-10-15 10:13:05 | | | |
| 2 | Jason Zhu | -4 word(s) | 2306 | 2021-10-19 04:10:21 | | | | |
| 3 | Kanako Miyano | -4 word(s) | 2302 | 2021-10-19 04:25:03 | | | | |
| 4 | Jason Zhu | -37 word(s) | 2265 | 2021-10-20 08:37:01 | | | | |
| 5 | Jason Zhu | -37 word(s) | 2265 | 2021-10-20 08:38:04 | | | | |
| 6 | Jason Zhu | Meta information modification | 2265 | 2021-10-26 11:52:08 | | | | |
| 7 | Jason Zhu | Meta information modification | 2265 | 2021-10-29 11:54:30 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Miyano, K. Oxytocin. Encyclopedia. Available online: https://encyclopedia.pub/entry/15113 (accessed on 22 July 2026).
Miyano K. Oxytocin. Encyclopedia. Available at: https://encyclopedia.pub/entry/15113. Accessed July 22, 2026.
Miyano, Kanako. "Oxytocin" Encyclopedia, https://encyclopedia.pub/entry/15113 (accessed July 22, 2026).
Miyano, K. (2021, October 19). Oxytocin. In Encyclopedia. https://encyclopedia.pub/entry/15113
Miyano, Kanako. "Oxytocin." Encyclopedia. Web. 19 October, 2021.
Copy Citation
Oxytocin (OT) influences various physiological functions such as uterine contractions, maternal/social behavior, and analgesia. OT acts as a positive allosteric modulator (PAM) for κ-opioid but not δ-opioid receptors and enhances κ-opioid receptor activity.
oxytocin
kappa-opioid receptor
positive allosteric modulator
1. Introduction
Opioid receptors (ORs), which belong to the class A family of G protein-coupled receptors (GPCRs), are mainly classified into three different types: μ-OR (MOR), δ-OR (DOR), and κ-OR (KOR) [1][2][3]. In addition to these three major receptors, the nociception/orphanin FR receptor was identified as the fourth member of the OR family [4]. Moreover, other opioid receptors, such as ζ-, ε-, λ-, and ι-ORs, have also been characterized [5]. The three classical ORs (MOR, DOR, and KOR) are primarily expressed in the spinal cord, brain stem, thalamus, and cortex, which together constitute the ascending pain transmission system and the descending inhibitory pain system [6]. Activation of these ORs decreases presynaptic transmitter release, hyperpolarization of postsynaptic elements, and disinhibition6. Endogenous opioid peptides, such as β-endorphin and dynorphin A, are related to the endogenous pain modulatory system [6]. Traditional opioid drugs, such as morphine, are used for acute and chronic cancer pain. However, these drugs often cause adverse effects, including respiratory depression, dependency, and tolerance; therefore, in-depth research has been performed on opioid cell signaling to overcome these adverse effects [7][8].
To overcome these adverse effects of the existing opioid drugs, research on G protein-biased agonists or positive allosteric modulators (PAMs) is being conducted to develop novel opioid drugs. G protein-biased agonists activate G protein-dependent signaling, but have a lesser effect on β-arrestin-dependent signaling.
PAMs bind to an allosteric site on the receptor, which is separate from the orthosteric site that binds an endogenous agonist, and then potentiates the affinity and/or efficacy of orthosteric agonists [9]. To date, many GPCRs have been found to be PAMs [10][11]. In the case of ORs, PAMs for MOR and for both MOR and DOR were identified in the 2010s [11][12]. These PAMs can only modulate the activity of ORs when the orthosteric agonists occupy the receptor, indicating that PAMs maintain spatial and temporal control of receptor signaling in vivo. Endogenous opioid peptides are released at locations in the brain or spinal cord where they are required, namely, where pain occurs [13]. Therefore, the use of PAMs could provide drugs with improved adverse effect profiles or lesser tolerance and dependence than orthosteric opioid receptor agonists. Therefore, G protein-biased PAMs for ORs are also potential candidates for analgesics with fewer adverse effects.
Oxytocin (OT), composed of nine amino acids, is synthesized in the paraventricular nucleus (PVN) and supraoptic nucleus of the hypothalamus. OT is secreted from the posterior pituitary, which has a wide range of physiological functions, such as uterine contractions, maternal/social behavior, and antistress effects [14][15][16]. Some studies have shown that OT alleviates pain in several brain regions and the spinal cord, but the molecular mechanism remains unclear [17][18][19]. It is presumed that multiple pathways are involved in the analgesic mechanism of OT, namely, OT receptor-dependent and OT receptor-independent pathways. The OT receptor-dependent pathway is a hypothalamospinal oxytocinergic pathway. PVN stimulation or OT administration activates presynaptic OT receptors superficially located in the dorsal horn (laminae I and II) and subsequently excites inhibitory GABAergic interneurons [20][21][22][23]. Activation of GABAergic interneurons presynaptically inhibits nociceptive nerves (Aδ-fiber and C-fiber). In a study involving the OT receptor-independent pathway, OT-induced analgesia was absent in vasopressin-1A receptor null mutant mice in the radiant heat paw-withdrawal test and von Frey test [24]. OT also attenuates pain via desensitization of transient receptor potential vanilloid 1 (TRPV1) [25]. Moreover, the endogenous opioid system is involved in OT-induced analgesia. Injection of OT into the periaqueductal gray was found to increase endogenous opioids, and the OT-induced antinociception was antagonized by the OR antagonist naloxone [26][27].
2. OT Alone Did Not Change Impedance (ΔZiec) in HEK293 Cells Stably Expressing Either Human DOR or KOR in the CellKeyTM Assay
To clarify the effects of OT on DORs and KORs, researchers performed the CellKeyTM assay to examine whether OT exerts an agonistic effect on DORs and KORs using DOR- and KOR-expressing HEK293 cells, respectively. The selective DOR agonist SNC80 and the selective KOR agonist U50488 increased ΔZiec in DOR- and KOR-expressing cells, respectively, in a dose-dependent manner; OT (10−10–10−5 M) when used alone did not affect ΔZiec in both DOR- and KOR-expressing cells (Figure 1A,B).
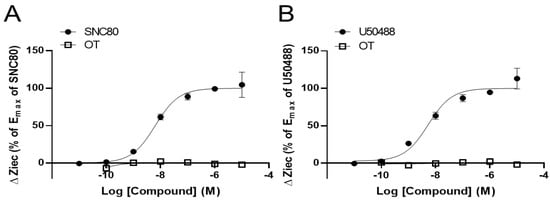
Figure 1. Oxytocin (OT) did not exert an agonistic effect on the human δ-opioid receptor (DOR) and κ-OR (KOR). HEK293 cells stably expressing DOR were treated with OT (10−10–10−5 M) or the selective DOR agonist SNC80 (10−11–10−5 M) (A). HEK293 cells stably expressing KOR were treated with OT (10−10–10−5 M) or the selective KOR agonist (-)-U50488H (U50488, 10−11–10−5 M) (B). The data were normalized by the Emax value of each selective agonist SNC80 (10−5 M; (A)) or U50488 (10−5 M; (B)) and are presented in terms of mean ± standard error of mean (SEM) values ((A); n = 3–4, (B); n = 3).
3. OT Enhanced ΔZiec Induced by KOR Agonists but Not DOR Agonists in HEK293 Cells Stably Expressing Human KOR or DOR, Respectively
OT (10−6 M) did not affect DOR activity induced by the DOR agonists SNC80 (10−11–10−5 M) and Leu- and Met-enkephalin (10−11–10−6 M) (Figure 2A–C). In contrast, OT (10−6 M) enhanced the Ziec induced by the KOR agonists U50488 (10−7–10−5 M) and dynorphin A (10−6–10−5 M) (Figure 2D,E).
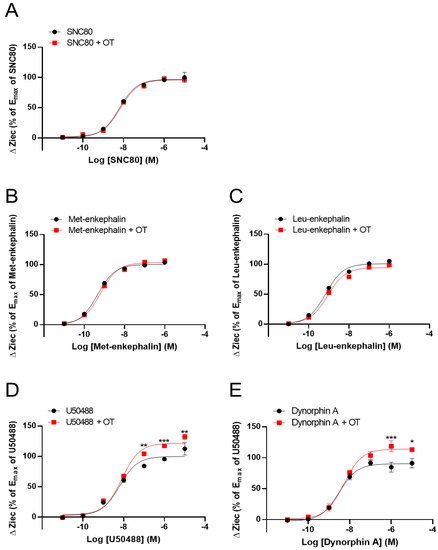
Figure 2. Effect of OT on the impedance (ΔZiec) induced by each KOR or DOR agonist in HEK293 cells stably expressing human KOR or DOR, respectively. Dose-response curves for SNC80- (A), Met-enkephalin- (B), and Leu-enkephalin- (C) induced ΔZiec in DOR-expressing cells, which were treated with or were not treated with 10−6 M OT. KOR-expressing cells were treated with U50488 (D) or dynorphin A (E) in the absence and presence of 10−6 M OT. The data were normalized by the Emax value of a selective DOR agonist (10−5 M SNC80) or a selective KOR agonist (10−5 M U50488) and are presented in terms of mean ± SEM values ((A); n = 5, (B); n = 3, (C); n = 3, (D); n = 4, (E); n = 3). Two-way ANOVA was performed, followed by Bonferroni’s test. * p < 0.05, ** p < 0.01, *** p < 0.001 compared to each agonist treatment alone.
It has been investigated the effect of OT on ΔZiec induced by opioid analgesics that selectively activate MORs over DORs or KORs. Morphine and fentanyl partially increased ΔZiec in both DOR- and KOR-expressing cells, indicating that these drugs were partial agonists for both DOR and KOR (Figure 3A–D). In DOR-expressing cells, OT (10−6 M) did not enhance ΔZiec induced by morphine (10−11–10−5 M) and fentanyl (10−12–10−6 M) (Figure 3A,B). In contrast, in KOR-expressing cells, the ΔZiec induced by morphine (10−5 M) but not fentanyl (10−12–10−6 M) was slightly enhanced by OT (Figure 3C,D). OT increased the Emax values of U50488 and dynorphin A (Table 1) and also increased the maximum morphine response (Figure 3C) without affecting the LogEC50 values in KOR-expressing cells (Table 1).
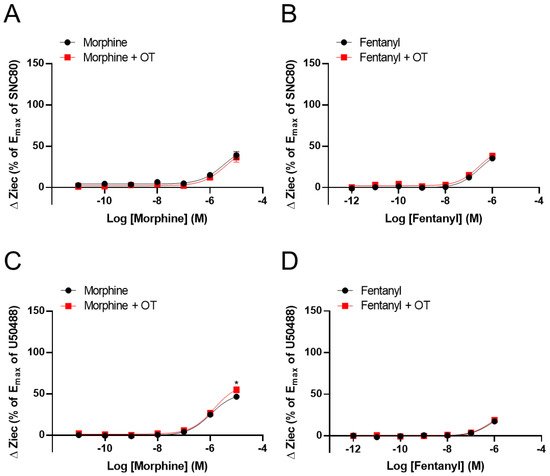
Figure 3. Effect of OT on ΔZiec induced by opioid analgesics in HEK293 cells stably expressing human DOR or KOR. Dose-response curves for morphine- (A) and fentanyl- (B) induced ΔZiec in the absence and presence of 10−6 M OT in DOR-expressing cells. KOR-expressing cells were treated with morphine (C) or fentanyl (D) in the absence and presence of 10−6 M OT. The data were normalized by the Emax value of a selective DOR agonist (10−5 M SNC80) or a selective KOR agonist (10−5 M U50488) and are presented in terms of mean ± SEM values (A); n = 4, (B); n = 3, (C); n = 4, (D); n = 4). Two-way ANOVA was performed, followed by Bonferroni’s test. * p < 0.05 compared to each agonist treatment alone.
Table 1. LogEC50 and Emax of each opioid in the CellKey assay, in the absence and presence of 10−6 M OT in HEK293 cells stably expressing human DOR or KOR.
| OR | Compounds | LogEC50 (M) | Emax (%) | ||
|---|---|---|---|---|---|
| Agonist | Agonist + OT | Agonist | Agonist + OT | ||
| DOR | SNC-80 | −8.20 ± 0.11 | −8.15 ± 0.06 | 100 ± 3.23 | 99.13 ± 1.77 |
| Met-enkephalin | −9.38 ± 0.08 | −9.34 ± 0.11 | 107.8 ± 2.33 | 112.65 ± 3.43 | |
| Leu-enkephalin | −9.23 ± 0.09 | −9.04 ± 0.12 | 108.4 ± 2.71 | 103.1 ± 3.59 | |
| Morphine | n.d. | n.d. | n.d. | n.d. | |
| Fentanyl | n.d. | n.d. | n.d. | n.d. | |
| KOR | U50488 | −8.20 ± 0.12 | −8.09 ± 0.07 | 100.0 ± 3.47 | 121.7 ± 2.75 ** |
| Dynorphin A | −8.51 ± 0.11 | −8.28 ± 0.09 | 90.3 ± 2.71 | 114.1 ± 2.97 ** | |
| Morphine | n.d. | n.d. | n.d. | n.d. | |
| Fentanyl | n.d. | n.d. | n.d. | n.d. | |
The effects of OT concentration dependency on KOR activity in KOR-expressing cells have been examined. OT enhanced the increase in ΔZiec induced by U50488 (10−6 M), dynorphin A (10−6 M), and morphine (10−5 M) in a concentration-dependent manner (Figure 4A–C).
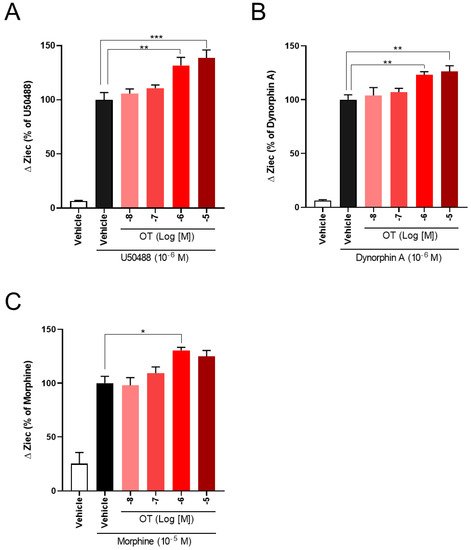
Figure 4. OT enhanced ΔZiec induced by KOR agonists in a concentration-dependent manner in HEK293 cells stably expressing human KOR. The cells were treated with 10−6 M U50488 (A), 10−6 M dynorphin A (B), or 10−5 M morphine (C). The data are presented in terms of mean ± SEM values ((A); n = 4, (B); n = 5, (C); n = 3–4). One-way ANOVA was performed, followed by Bonferroni’s test, * p < 0.05, ** p < 0.01, *** p < 0.001 compared to each agonist alone.
4. OT Enhanced Inhibition of Intracellular cAMP Induced by KOR Agonists in HEK293 Cells Stably Coexpressing Both Human KOR and Glosensor 22F Protein in the GlosensorTM cAMP Assay
The CellkeyTM assay detects the activation of GPCRs as whole-cell responses on the basis of changes in electrical impedance (ΔZ) [29]. Two of the most important GPCR signaling pathways are the G protein-dependent signaling and the β-arrestin-dependent signaling pathway. DOR and KOR are Gi/o protein-coupled GPCRs; therefore, their activation leads to the inhibition of adenylyl cyclase and decreased intracellular cAMP. Thus, researchers analyzed the effects of OT on KOR-mediated downstream signaling using the GlosensorTM cAMP assay. U50488 (10−6 M), dynorphin A (10−6 M), and morphine (10−5 M) considerably suppressed intracellular cAMP production induced by forskolin (vehicle), adenylyl cyclase activator [30], and the inhibition was enhanced by treatment with 10−6 M OT (Figure 5A–C).
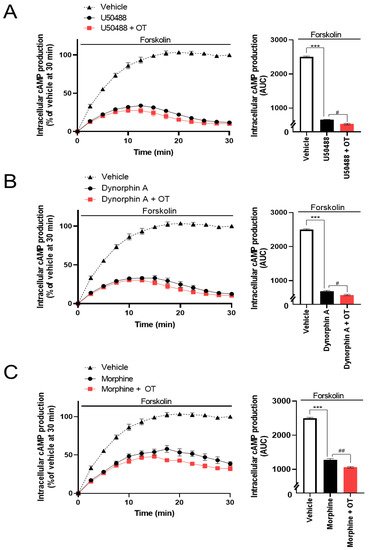
Figure 5. Effects of OT on inhibition of intracellular cAMP production induced by each KOR agonist in HEK293 cells stably coexpressing human KOR and Glosensor 22F protein. The cells were pretreated with 10−6 M U50488 (A), 10−6 dynorphin A (B), or 10−5 M of morphine (C) in the absence and presence of 10−6 M OT for 10 min and treated with forskolin (3 µM). The data are presented in terms of mean ± SEM values ((A); n = 3, (B); n = 3, (C); n = 3). Two-way ANOVA was performed, followed by Bonferroni’s test, *** p < 0.001 compared to vehicle. # p < 0.05 and ## p < 0.01 compared to each agonist alone.
5. OT Did Not Potentiate the KOR Internalization Induced by KOR Agonists in HEK293 Cells Stably Expressing the Human KOR Fused Halotag® in the Internalization Assay
To assess the effects of OT on β-arrestin-dependent signaling mediated by KOR, we performed an internalization assay with the Halotag® pH Sensor Ligand to quantify KOR internalization, as previously reported by Manabe et al. for MOR37. In this internalization assay, Halotag®-fused KOR bound to Halotag® pH Sensor Ligand in an acidic environment (e.g., endosomes), fluoresced as red spots. Treatment with OT alone for 120 min did not increase the quantity of red spots (Figure 6A). U50488 (10−6 M) and dynorphin A (10−6 M) caused KOR internalization; however, very few red spots were noted on treatment with morphine (10−5 M), until 120 min after drug injection (Figure 6B–D). In contrast to the cAMP assay findings, OT did not significantly affect these phenomena in this assay (Figure 6B–D).
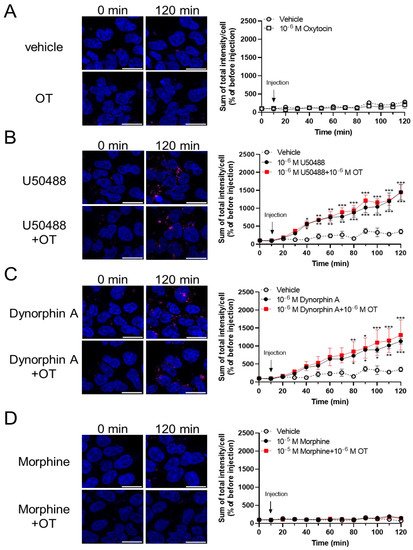
Figure 6. OT did not enhance internalization of KOR induced by dynorphin A and the U50488 agonist in HEK293 cells stably expressing HaloTag®-fused human KOR. Cells were dyed with a 0.5 mM pH sensor ligand and Hoechst 33342. After washing, the cells were treated with vehicle (A), U50488 (10−5 M), (B), dynorphin A (10−6 M) (C), or morphine (10−5 M) (D) in the absence and presence of 10−6 M OT. Time-response curves were calculated as the sum of total red fluorescence intensity per cell and were normalized to those before injection (at 0 min). The data are presented in terms of mean ± SEM values ((A); n = 4, (B); n = 3, (C); n = 3, (D); n = 3). Two-way ANOVA was performed, followed by Bonferroni’s test, * p < 0.05, ** p < 0.01, *** p < 0.001 compared to vehicle. Scale bar = 20 µm.
6. OT Did Not Affect [3H]Diprenorphine Binding to CHO Cells Stably Expressing Human KOR in the Radioligand Competitive OR Binding Assay
While some PAMs potentiate the binding affinity of agonists for the receptors [31][32], OT did not affect the binding affinity of MOR [28]. To determine whether OT binds to the KOR orthosteric binding sites or enhances the binding affinity of KOR agonists for KOR, researchers performed a radioligand competitive OR binding assay with [3H]diprenorphine. OT, when used alone (10−11–10−5 M), did not inhibit [3H]diprenorphine binding to KOR (Figure 7A). Furthermore, there was no difference in the LogIC50 and LogKi values between dynorphin A and dynorphin A+OT, or U50488 and U50488+OT, respectively (Figure 7B,C, Table 2).
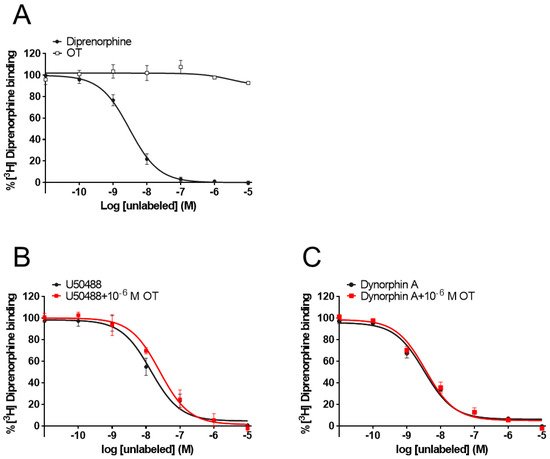
Figure 7. OT neither bound to orthosteric binding sites nor enhanced the binding affinity of KOR agonists for KOR in membranes prepared from CHO cells stably expressing human KOR. Membranes prepared from KOR-expressing cells were treated with OT or dynorphin A alone (A). U50488 (10−11–10−5 M, (B) or dynorphin A (10−11–10−5 M, (C) were treated with or without 10−6 M OT. The data are presented in terms of the mean ± SEM values ((A); n = 3, (B); n = 3, (C); n = 3).
Table 1. LogEC50 and Emax of each opioid in the CellKey assay, in the absence and presence of 10−6 M OT in HEK293 cells stably expressing human DOR or KOR.
| OR | Compounds | LogEC50 (M) | Emax (%) | ||
|---|---|---|---|---|---|
| Agonist | Agonist + OT | Agonist | Agonist + OT | ||
| DOR | SNC-80 | −8.20 ± 0.11 | −8.15 ± 0.06 | 100 ± 3.23 | 99.13 ± 1.77 |
| Met-enkephalin | −9.38 ± 0.08 | −9.34 ± 0.11 | 107.8 ± 2.33 | 112.65 ± 3.43 | |
| Leu-enkephalin | −9.23 ± 0.09 | −9.04 ± 0.12 | 108.4 ± 2.71 | 103.1 ± 3.59 | |
| Morphine | n.d. | n.d. | n.d. | n.d. | |
| Fentanyl | n.d. | n.d. | n.d. | n.d. | |
| KOR | U50488 | −8.20 ± 0.12 | −8.09 ± 0.07 | 100.0 ± 3.47 | 121.7 ± 2.75 ** |
| Dynorphin A | −8.51 ± 0.11 | −8.28 ± 0.09 | 90.3 ± 2.71 | 114.1 ± 2.97 ** | |
| Morphine | n.d. | n.d. | n.d. | n.d. | |
| Fentanyl | n.d. | n.d. | n.d. | n.d. | |
7. Conclusion
In conclusion, OT has KOR-PAM activity but does not have DOR-PAM activity. OT enhances G protein signaling without affecting β-arrestin signaling in KOR, indicating that OT has potential as a G protein-biased PAM of KOR. Thus, OT might act as a natural analgesic against pain via modulation of OR signaling.
References
- Chen, Y.; Mestek, A.; Liu, J.; Hurley, J.A.; Yu, L. Molecular cloning and functional expression of a mu-opioid receptor from rat brain. Mol. Pharmacol. 1993, 44, 8–12.
- Kieffer, B.L.; Befort, K.; Gaveriaux-Ruff, C.; Hirth, C.G. The delta-opioid receptor: Isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. USA 1992, 89, 12048–12052.
- Minami, M.; Toya, T.; Katao, Y.; Maekawa, K.; Nakamura, S.; Onogi, T.; Kaneko, S.; Satoh, M. Cloning and expression of a cDNA for the rat kappa-opioid receptor. FEBS Lett. 1993, 329, 291–295.
- Meunier, J.C. Utilizing functional genomics to identify new pain treatments: The example of nociceptin. Am. J. Pharm. Genom. 2003, 3, 117–130.
- Brott, N.R.; Peterson, E.; Cascella, M. Opioid, Risk Tool. In StatPearls; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021.
- Inturrisi, C.E. Clinical pharmacology of opioids for pain. Clin. J. Pain 2002, 18, S3–S13.
- Stein, C. Opioid receptors. Annu. Rev. Med. 2016, 67, 433–451.
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520.
- Conn, P.J.; Christopoulos, A.; Lindsley, C.W. Allosteric modulators of GPCRs: A novel approach for the treatment of CNS disorders. Nat. Rev. Drug. Discov. 2009, 8, 41–54.
- Gentry, P.R.; Sexton, P.M.; Christopoulos, A. Novel allosteric modulators of G protein-coupled receptors. J. Biol. Chem. 2015, 290, 19478–19488.
- Burford, N.T.; Clark, M.J.; Wehrman, T.S.; Gerritz, S.W.; Banks, M.; O’Connell, J.; Traynor, J.R.; Alt, A. Discovery of positive allosteric modulators and silent allosteric modulators of the μ-opioid receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 10830–10835.
- Burford, N.T.; Livingston, K.E.; Canals, M.; Ryan, M.R.; Budenholzer, L.M.; Han, Y.; Shang, Y.; Herbst, J.J.; O’Connell, J.; Banks, M.; et al. Discovery, synthesis, and molecular pharmacology of selective positive allosteric modulators of the δ-opioid receptor. J. Med. Chem. 2015, 58, 4220–4229.
- Rasmussen, N.A.; Farr, L.A. Beta-endorphin response to an acute pain stimulus. J. Neurosci. Methods 2009, 177, 285–288.
- Gimpl, G.; Fahrenholz, F. The oxytocin receptor system: Structure, function, and regulation. Physiol. Rev. 2001, 81, 629–683.
- Lee, S.Y.; Park, S.H.; Chung, C.; Kim, J.J.; Choi, S.Y.; Han, J.S. Oxytocin protects hippocampal memory and plasticity from uncontrollable stress. Sci. Rep. 2015, 5, 18540.
- Yamasue, H.; Yee, J.R.; Hurlemann, R.; Rilling, J.K.; Chen, F.S.; Meyer-Lindenberg, A.; Tost, H. Integrative approaches utilizing oxytocin to enhance prosocial behavior: From animal and human social behavior to autistic social dysfunction. J. Neurosci. 2012, 32, 14109–14117.
- Lundeberg, T.; Meister, B.; Björkstrand, E.; Uvnäs-Moberg, K. Oxytocin modulates the effects of galanin in carrageenan-induced hyperalgesia in rats. Brain Res. 1993, 608, 181–185.
- Petersson, M.; Alster, P.; Lundeberg, T.; Uvnäs-Moberg, K. Oxytocin increases nociceptive thresholds in a long-term perspective in female and male rats. Neurosci. Lett. 1996, 212, 87–90.
- Yang, J.; Yang, Y.; Chen, J.M.; Liu, W.Y.; Wang, C.H.; Lin, B.C. Central oxytocin enhances antinociception in the rat. Peptides 2007, 28, 1113–1119.
- Breton, J.D.; Veinante, P.; Uhl-Bronner, S.; Vergnano, A.M.; Freund-Mercier, M.J.; Schlichter, R.; Poisbeau, P. Oxytocin-induced antinociception in the spinal cord is mediated by a subpopulation of glutamatergic neurons in lamina I-II which amplify GABAergic inhibition. Mol. Pain 2008, 4, 19.
- Condés-Lara, M.; Rojas-Piloni, G.; Martínez-Lorenzana, G.; Rodríguez-Jiménez, J.; López Hidalgo, M.; Freund-Mercier, M.J. Paraventricular hypothalamic influences on spinal nociceptive processing. Brain Res. 2006, 1081, 126–137.
- Rojas-Piloni, G.; Martínez-Lorenzana, G.; DelaTorre, S.; Condés-Lara, M. Nociceptive spinothalamic tract and postsynaptic dorsal column neurons are modulated by paraventricular hypothalamic activation. Eur. J. Neurosci. 2008, 28, 546–558.
- Rojas-Piloni, G.; Mejía-Rodríguez, R.; Martínez-Lorenzana, G.; Condés-Lara, M. Oxytocin, but not vassopressin, modulates nociceptive responses in dorsal horn neurons. Neurosci. Lett. 2010, 476, 32–35.
- Schorscher-Petcu, A.; Sotocinal, S.; Ciura, S.; Dupré, A.; Ritchie, J.; Sorge, R.E.; Crawley, J.N.; Hu, S.B.; Nishimori, K.; Young, L.J.; et al. Oxytocin-induced analgesia and scratching are mediated by the vasopressin-1A receptor in the mouse. J. Neurosci. 2010, 30, 8274–8284.
- Nersesyan, Y.; Demirkhanyan, L.; Cabezas-Bratesco, D.; Oakes, V.; Kusuda, R.; Dawson, T.; Sun, X.; Cao, C.; Cohen, A.M.; Chelluboina, B.; et al. Oxytocin modulates nociception as an agonist of pain-sensing TRPV1. Cell Rep. 2017, 21, 1681–1691.
- Yang, J.; Liang, J.Y.; Li, P.; Pan, Y.J.; Qiu, P.Y.; Zhang, J.; Hao, F.; Wang, D.X. Oxytocin in the periaqueductal gray participates in pain modulation in the rat by influencing endogenous opiate peptides. Peptides 2011, 32, 1255–1261.
- Taati, M.; Tamaddonfard, E. Ventrolateral orbital cortex oxytocin attenuates neuropathic pain through periaqueductal gray opioid receptor. Pharmacol. Rep. 2018, 70, 577–583.
- Meguro, Y.; Miyano, K.; Hirayama, S.; Yoshida, Y.; Ishibashi, N.; Ogino, T.; Fujii, Y.; Manabe, S.; Eto, M.; Nonaka, M.; et al. Neuropeptide oxytocin enhances μ opioid receptor signaling as a positive allosteric modulator. J. Pharmacol. Sci. 2018, 137, 67–75.
- Miyano, K.; Sudo, Y.; Yokoyama, A.; Hisaoka-Nakashima, K.; Morioka, N.; Takebayashi, M.; Nakata, Y.; Higami, Y.; Uezono, Y. History of the G protein-coupled receptor (GPCR) assays from traditional to a state-of-the-art biosensor assay. J. Pharmacol. Sci. 2014, 126, 302–309.
- Uezono, Y.; Bradley, J.; Min, C.; McCarty, N.A.; Quick, M.; Riordan, J.R.; Chavkin, C.; Zinn, K.; Lester, H.A.; Davidson, N. Receptors that couple to 2 classes of G proteins increase cAMP and activate CFTR expressed in Xenopus oocytes. Recept. Channels 1993, 1, 233–241.
- Bisignano, P.; Burford, N.T.; Shang, Y.; Marlow, B.; Livingston, K.E.; Fenton, A.M.; Rockwell, K.; Budenholzer, L.; Traynor, J.R.; Gerritz, S.W.; et al. Ligand-Based discovery of a new scaffold for allosteric modulation of the μ-opioid receptor. J. Chem. Inf. Model. 2015, 55, 1836–1843.
- Koole, C.; Wootten, D.; Simms, J.; Valant, C.; Sridhar, R.; Woodman, O.L.; Miller, L.J.; Summers, R.J.; Christopoulos, A.; Sexton, P.M. Allosteric ligands of the glucagon-like peptide 1 receptor (GLP-1R) differentially modulate endogenous and exogenous peptide responses in a pathway-selective manner: Implications for drug screening. Mol. Pharmacol. 2010, 78, 456–465.
More
Information
Subjects:
Allergy
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Entry Collection:
Peptides for Health Benefits
Revisions:
7 times
(View History)
Update Date:
29 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No