Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Miguel A. Alonso | + 3824 word(s) | 3824 | 2021-10-09 07:43:59 | | | |
| 2 | Bruce Ren | Meta information modification | 3824 | 2021-10-18 03:23:41 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Alonso, M.A. Formins in Human Monogenic Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/15075 (accessed on 24 July 2026).
Alonso MA. Formins in Human Monogenic Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/15075. Accessed July 24, 2026.
Alonso, Miguel A.. "Formins in Human Monogenic Disease" Encyclopedia, https://encyclopedia.pub/entry/15075 (accessed July 24, 2026).
Alonso, M.A. (2021, October 16). Formins in Human Monogenic Disease. In Encyclopedia. https://encyclopedia.pub/entry/15075
Alonso, Miguel A.. "Formins in Human Monogenic Disease." Encyclopedia. Web. 16 October, 2021.
Copy Citation
Almost 25 years have passed since a mutation of a formin gene, DIAPH1, was identified as being responsible for a human inherited disorder: a form of sensorineural hearing loss. Since then, our knowledge of the links between formins and disease has deepened considerably. Mutations of DIAPH1 and six other formin genes (DAAM2, DIAPH2, DIAPH3, FMN2, INF2 and FHOD3) have been identified as the genetic cause of a variety of inherited human disorders, including intellectual disability, renal disease, peripheral neuropathy, thrombocytopenia, primary ovarian insufficiency, hearing loss and cardiomyopathy.
formins
genetic disease
gene variants
developmental defects
actin
1. Introduction
The human formin family consists of fifteen members (Figure 1A), divided into seven subfamilies [1], many of which are co-expressed in many tissues. Formins are involved in the polymerization of monomeric actin into linear filaments [2][3]. All formins possess two characteristic domains: a formin homology (FH) 2 domain, which catalyzes actin polymerization, and an FH1 domain, which binds profilin to provide monomeric actin to the FH2 domain. The other regions and domains can differ between formin subfamilies and are involved in regulatory mechanisms or specific interactions with other proteins. In addition to regulating the actin cytoskeleton, formins bind to microtubules through the FH2 domain and regulate the acetylation and stability of microtubules, and their alignment with actin filaments [4][5].
Members of the human Diaphanous-related formin subfamily, which includes Diaphanous homolog (DIAPH) 1-3, are regulated through the interaction of the Diaphanous inhibitory domain (DID) at the N-terminal end and the Diaphanous autoregulatory domain (DAD) at the C-terminal region [1]. The transition between closed/inactive and open/active states is mediated by the interaction of the Rho-family GTPases with the DID, which releases its interaction with the DAD (Figure 1B). Other formins with similar regulation are Disheveled-associated activators of morphogenesis (DAAM) 1 and 2, formin-like (FMNL) 1-3, and FH1/FH2 domain-containing (FHOD) 1 and 3.
The mutation of some of the formin genes causes monogenic disorders, as is the case of DIAPH1, which was the first formin gene found to be linked to a human Mendelian disorder [6]. Alteration of seven formins genes (Figure 2) have been acknowledged to date by the Online Mendelian Inheritance in Man (OMIM®, McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University (Baltimore, MD, USA), https://omim.org; accessed on 20 September 2021) as meeting the criteria for consideration as a primary cause of human monogenic disorders [7][8]: DIAPH1-3 [6][9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][28][29][30][31][32][33][34][35][36], DAMM2 [37], FORMIN2 [38][39][40][41][42] (FMN2) , INVERTED FORMIN 2 (INF2) [43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94] and FHOD3 [95][96][97][98][99][100] . The mutations or dysregulation of the other formins have not been demonstrated to be the primary cause of the phenotype, although they probably contribute to it [101][102]. Mutant formins can alter specific organs by affecting the functioning of specific types of cell (Figure 3A,B).
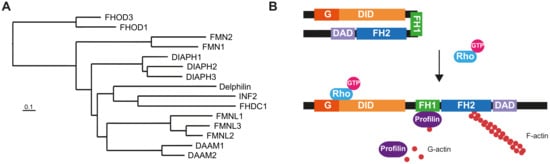
Figure 1. The human formin family. (A) Tree of human formins. The FH2 domain sequence of the formins was aligned with BLAST and the alignment was used to construct the tree [103]. The UniProt accession numbers of the corresponding sequences were: DIAPH1 (O60610), DIAPH2 (O60879), DIAPH3 (Q9NSV4), DAAM1 (Q9Y4D1), DAAM2 (Q86T65), FMNL1 (O95466), FMNL2 (Q96PY5), FMNL3 (Q8IVF7), FHOD1 (Q9Y613), FHOD3 (Q2V2M9), FMN1 (Q68DA7), FMN2 (Q9NZ56), INF2 (Q27J81), FHDC1 (Q9C0D6) and Delphilin (A4D2P6). (B) Structure and regulation of Diaphanous-related formins. The interaction of the DID and the DAD maintains the formin in a closed, inactive conformation. The binding of a specific GTP-loaded Rho GTPase to the N-terminal region of the formin opens the molecule, rendering it in its active form. The FH1 domain recruits profilin, which feeds the FH2 domain with G-actin to form the actin filaments. The illustrated molecules are not drawn to scale.
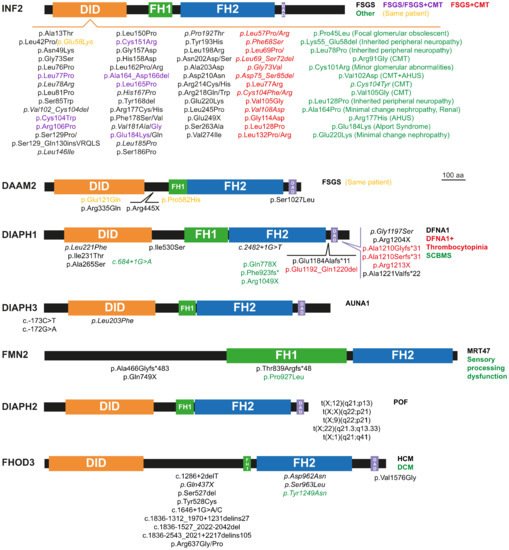
Figure 2. Pathogenic mutations of the formins causing monogenic disorders. Depending on the specific mutation, some formins produce different disorders. In these cases, we used the colors, as indicated, to refer to each of the diseases and the corresponding mutations. *, stop codon. The mutations without reported familial studies are indicated in italics.
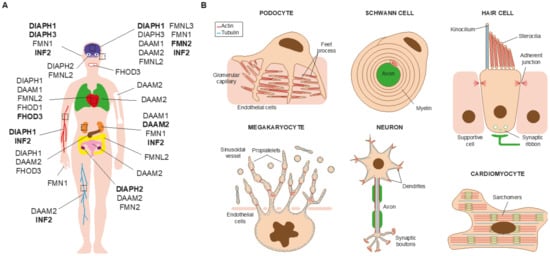
Figure 3. Some of the organs and cell types affected by formin alterations. (A) The formins involved in human disorders and the affected organs and systems are indicated in the schematic of the human body. Those causing monogenic disorders are highlighted in bold. (B) Some of the affected cell types. Their most characteristic structures are indicated.
2. Monogenic Disorders Caused by Formin Mutation
2.1. Nephrotic Syndrome and Charcot-Marie-Tooth Disease
Blood filtration and the concentration of metabolic waste into urine take place in the renal glomeruli. Podocytes are terminally differentiated cells that wrap around endothelial cells of glomerular capillaries by means of elaborate projections known as foot processes (Figure 3B). The contact between two of these processes forms a slit diaphragm, which is the structure responsible for blood filtration [104]. Deficient blood filtration causes nephrotic syndrome, which is characterized by proteinuria, hypoalbulinemia, hyperlipidemia, and edema, and can end in renal failure.
Focal segmental glomerulosclerosis (FSGS) refers to a histological lesion of scarred appearance present in localized regions of some, but not all, glomeruli [105]. The INF2 gene is the formin for which the greatest number of pathogenic mutations has been described (Figure 2 and Supplementary Table S8) [106]. INF2 pathogenic mutations are autosomal dominant and produce FSGS (FSGS5, MIM: 613237) [43], which causes steroid-resistant nephrotic syndrome [107][108]. Depending on the specific mutation, FSGS co-occurs (or not) with Charcot–Marie–Tooth disease (CMTDIE, MIM: 614455) [48], which is a neuropathy affecting the functioning of the peripheral nerves that produces progressive distal muscle weakness [109]. All the INF2 disease-related mutations localize to the DID and the great majority are of the missense type. Genomic-wide screening (GWS) and whole-exome sequencing (WES) analyses in patients with renal disease identified a number of variants outside the INF2 DID [106] but, with the exception of a case of FSGS combined with CMT with a deletion in the DAD [94], it is not clear whether these variants are related to the pathogenic condition.
An in silico analysis of the effect of the pathogenic mutations indicates that they have a destabilizing structural effect in the DID [106]. This destabilization might affect the interaction of the DID with the DAD or with regulatory proteins [110][111], and results in gain-of-function of the actin polymerization activity of INF2. The case of a patient with combined FSGS and CMT has been described, in which a complete duplication of the INF2 gene occurred, which represents further evidence of a gain-of-function phenotype in INF2-linked disease [93]. It is of note that the mutations causing combined FSGS and CMT are generally more destabilizing than those producing only FSGS, and that these two types of mutation distribute in the DID in a different manner, with the former being concentrated in the N-terminal half of the DID, whereas those causing only FSGS are distributed throughout the DID [106].
FSGS patients suffer a progressive loss of podocytes, which decreases the filtration capacity of the kidney. INF2-linked FSGS starts to become clinically relevant in adolescence or adulthood, causing glomerular dysfunction [107][108]. It is still not clear how INF2 mutations affect podocytes but, consistent with the enhanced actin polymerization activity of the pathogenic INF2 mutants [110], aberrant actin bundles have been observed in a renal biopsy of an affected patient [43]. Knock-in mice expressing the most common mutation, p.Arg218Gln, exhibit no apparent alteration in podocyte structure unless they are exposed to acute kidney injury [112]. This finding is consistent with the degenerative nature of INF2-related disease and suggests that FSGS might be the result of repeated kidney insults in individuals in which INF2 mutation makes them more prone to developing the disease. In addition to FSGS, INF2 mutations have been found to contribute to, or be responsible for, other kidney conditions (Figure 2 and Supplementary Table S8). In patients with combined FSGS and CMT, CMT symptoms appear in childhood, and renal damage appears earlier in life than in patients with only FSGS. In the cases with CMT, pathogenic INF2 affects Schwann cell polarization (Figure 3B), leading to abnormal myelin formation and/or maintenance [113][114]. The manifestation of FSGS alone is common in individuals with pathogenic INF2, but only one case of CMT has been described to date that makes the absence of accompanying renal disease explicit [59].
Recessive mutations of DAAM2 (Figure 2 and Supplementary Table S6), have recently been involved in nephrotic syndrome, type 24 (NPHS24, MIM: 606627) [37]. All the affected individuals presented FSGS with no extra-renal manifestations. The mutations were found in homozygosity in three individuals from consanguineous families, and in one individual with two different missense mutations from an outbred family. The missense mutations map to the region encoding the DID, the FH1 or the DAD, whereas the nonsense mutation maps immediately downstream of the DID and generates a truncated DAAM2 protein. The mutations at the DID and DAD appear to cause increased autoinhibition and, consequently, loss-of-function of actin polymerization activity. DAAM2, which is expressed by podocytes, colocalizes and associates with INF2 [37], suggesting the existence of crosstalk between the two formins that, given the link between INF2 and renal disease, may explain the renal damage caused by pathogenic DAAM2. Other formins might be involved in other kidney disorders. For instance, Fmn1 has recently been identified as a candidate modifier gene in X-linked Alport syndrome in mice, which is a genetic disease characterized by hearing loss, hematuria and, eventually, renal failure [115].
2.2. Hearing Loss
Hearing depends on the correct mechanostransduction of sound vibrations into electrical signals. This takes place in the organ of Corti, which is located in the cochlea in the inner ear. The cells responsible for this process are sensory epithelial cells, known as hair cells (Figure 3B), which possess dozens of stereocilia on their apical surface, formed of bundles of actin filaments [116]. Outer hair cells amplify the signal, and the perturbation activates the opening of ion channels at the stereocilia tips of inner hair cells, depolarizing the plasma membrane. This perturbation is subsequently transmitted by neurotransmitters released at the synaptic ribbon between the basolateral surface of hair cells and the auditory nerve. This generates electrical impulses in the latter that are transmitted to the brain, where they are decoded and analyzed in the auditory cortex. Given the importance of actin in the architecture of stereocilia, mutations in actin, actin-binding proteins, and the machinery involved in actin filament formation and function, including formins, can all cause hearing loss [117].
Sensorineural hearing loss is caused by dysfunction of the inner ear or the auditory nerve. Mutations in DIAPH1 produce deafness, autosomal dominant 1 (DFNA1, MIM: 124900). In this disorder, auditory loss generally starts during the first decade of life, although there are cases with intrafamilial variability. Since the identification of a mutation in DIAPH1 as the cause of sensorineural hearing loss in a large Costa Rican family [6], more families with a dominant pedigree caused by DIAPH1 mutation have been described elsewhere in the world [11][12][13][18][19][21][22][23][24][25][26][27]. Affected individuals present frameshift or nonsense mutations or deletions near the DAD. These types of mutation create truncated forms of DIAPH1 that lack different segments of the carboxyl-terminal region of the molecule. In addition, more recently, missense mutations have been described at the DID and the coiled-coil downstream region and FH2 domain (Figure 2 and Supplementary Table S3). The pathogenic p.Arg1204X mutation [21] results in early termination immediately before a basic amino acid motif (RRKR1204–1207) present at the DAD C-terminus, which is important for the interaction with the DID [118]. This mutation partially relieves the autoinhibitory DID-DAD interaction, resulting in a mildly constitutive active molecule [21]. It is likely that this also occurs with other truncation mutations mapping around this site and with the missense mutations in the DID [119][120]. The DIAPH1 gene mouse homolog, mDia1, is expressed in the organ of Corti during and after cochlear maturation, and localizes at the apical junctional complexes between the supporting cells and the hair cells [121]. As further evidence that hearing loss caused by DIAPH1 mutations is due to gain- and not to loss-of-function, hearing progressively deteriorates in transgenic mice overexpressing wild-type mDia1 [121], whereas the hearing function of the mDia1 knock-out (KO) mice is not different from that of control mice [21]. The hearing defect in mice overexpressing mDia1 is associated with gradual loss of hair cells and the appearance of sparse and short or fused stereocilia cells [121]. A similar phenotype was observed in transgenic mice expressing the human Arg1204X mutant [21]. Increased gene dosage of DIAPH1 has been documented in several cases of sporadic sensorineural hearing loss in humans [28]. These findings are further evidence that deafness-associated mutations of DIAPH1 cause disease by increasing actin polymerization activity, which causes the disorganization and dysfunction of stereocilia.
Auditory neuropathy, autosomal dominant, 1 (AUNA1, MIM: 609129) is characterized by abnormal or absent auditory brainstem responses but preserved cochlear outer hair cell function. A mutation (c.-172G > A) in a highly conserved GC element at the exon encoding the 5′ untranslated region of DIAPH3, was the first to be described as being involved in AUNA1. This mutation, which is probably of the gain-of-function type, results in 2- to 3-fold overexpression of DIAPH3 mRNA and 1.5-fold overexpression of DIAPH3 protein levels. Consistent with increased levels of DIAPH3 as the cause of the auditory alterations, flies expressing a constitutively active form of Diaphanous, which encodes the sole Diaphanous-related formin in Drosophila, show an impaired response to sound [36]. Two reports found that mice overexpressing mouse mDia2, the murine homolog of human DIAPH3, present progressive impairment of inner hair cell stereocilia, whereas outer hair cells stereocilia and function were not generally affected in the specific mouse lines studied [122][123]. A reduction in the number of ribbon synapses was observed in one study [122], but not in the other [123]. Consistent with the role of formins in regulating microtubule dynamics, the microtubule meshwork undergoes aberrant targeting to the apical aspect of inner hair cells in transgenic mDia2 mice [123], probably contributing to stereocilia collapse. These mice also present early mortality due to cardiac defects, but no similar effect has yet been found in humans. In addition to the c.-172G > A mutation, other mutations causing AUNA1 have been described at the 5′ untranslated region of DIAPH3 mRNA [35] and the DID of DIAPH3 [15] (Figure 2, Supplementary Table S5). Missense variants mapping to the DIAPH3 FH2 domain have also been found in patients with auditory neuropathy spectrum disorders [15][124], but it is not clear whether there they are pathogenic or simply rare variants.
In the case of INF2 mutations associated with combined CMT plus FSGS, but not with FSGS alone, some of the patients also experience hearing loss [48][49][51][57]. Since INF2 mutations causing CMT affect peripheral nerve myelinization [113], auditory nerve damage is probably the cause of the hearing impairment, although hair cell stereocilia may also be affected, as in cases of DIAPH1 and DIAPH3 mutations.
2.3. Thrombocytopenia
The cell precursors of platelets, megakaryocytes, form extensions known as proplatelets, from which platelets are released into the circulatory system [125]. Macrothrombocytopenia is characterized by enlarged and reduced numbers of circulating platelets that can lead to inadequate clot formation and an increased risk of bleeding [125]. Platelet production begins with the extension of long membrane protrusions that are elongated by microtubule bundles to form proplatelet processes (Figure 3B). Amplification of the number of processes, which occurs by repeated bending and bifurcation, depends on actin filament formation [126][127][128]. It is controversial whether actin/microtubule crosstalk-induced proplatelet formation [126] or membrane budding without requiring proplatelet formation is the main mechanism of platelet formation in vivo [129].
Long after the discovery of the DIAPH1 mutation as the cause of DFNA1, affected individuals were found to present asymptomatic thrombocytopenia and, sometimes, asymptomatic mild neutropenia (Figure 2, Supplementary Table S3), which consists of abnormally low levels of neutrophils in the blood. The reduced platelet levels in these patients, the high content of polymerized actin, and the altered microtubule organization and stability observed in the platelets [27] are consistent with the requirement of DIAPH1 for proper proplatelet formation [130][131], and with previous works showing that DIAPH1 coordinates microtubules and the actin cytoskeleton [132][133]. It is of note that a moderate increase in the expression of DIAPH1 could be responsible for the thrombocytopenia associated with diseases caused by mutation in other genes, as may be the case for Roifman syndrome [134]. This is a rare, inherited disease (MIM: 616651) characterized by growth retardation, cognitive delays, skeletal malformations and immunodeficiency, and caused by mutation of the small non-coding RNA gene RNU4TAC [135].
Atypical hemolytic uremic syndrome (AHUS) is characterized by acute renal failure, thrombocytopenia, and microangiopathic hemolytic anemia (loss of blood cells through destruction). Two mutations in the DID of INF2 cause thrombocytopenia in the context of familial AHUS with (p.Val102Asp) or without (p.Arg177His) associated CMT [60]. In AHUS, the thrombocytopenia is due to platelet activation and consumption associated with blood cell destruction, rather than to an alteration in platelet production.
2.4. Microcephaly and Intellectual Disability
According to the Human Protein Atlas (http://www.proteinatlas.org; accessed on 30 August 2021), and consistent with the analyses of mouse brain [136], all the formins are expressed throughout the brain, generally with low regional specificity (Supplementary Table S2). Mutations of DIAPH1, FMN2, and INF2 are associated, to varying degrees, with intellectual disability and neurodevelopmental disorders [137].
DIAPH1 is expressed in neuronal progenitors during brain development [14]. Specific mutations of DIAPH1 cause seizures, cortical blindness (vision loss due to a damage or malfunction in the part of the brain cortex responsible for processing visual information), and microcephaly syndrome (SCBMS, MIM: 616632) (Figure 2, Supplementary Table S3). In contrast to DFNA1-related mutations, SCBMS-associated DIAPH1 mutations are generally of the nonsense type that affects the FH2 domain, are found in homozygosity, and are inherited with an autosomal recessive pattern, suggesting that they produce loss-of-function of DIAPH1 activity [10][14][16][17]. mDia1 KO mice are not microcephalic but, instead, some mice present unilateral dilatation of the ventricles, indicating that the effect of these mutations is species-specific [14]. Unlike mDia1 KO mice, mDia2 KO mice present microcephaly and also hydrocephalus (accumulation of cerebrospinal fluid within the brain) [138]. This phenotype seems to be due to incorrect spindle assembly checkpoint regulation in cortical progenitor cells, causing massive loss of cortical progenitor cells, with the subsequent depletion of neurons [138]. mDia1 and mDia3 double-KO mice present hydrocephalus, but not microcephaly, due to the formation of a periventricular dysplastic (abnormal) mass during brain development [139]. The alteration of the actin cytoskeleton affecting the adherens junctions and progenitors’ polarity seems to be the cause of the ectopic proliferation of neural stem cells in the double-KO mice. In addition to the characteristic SCBMS symptoms, some patients present pathologies related to immunodeficiency, such as recurrent infections, especially respiratory, bronchiectasis (enlargement of parts of the airways of the lung) and lymphoma [10][14][16]. Given that (i) mDia1 KO mice show defects in T cell migration and activation [140][141], (ii) DIAPH1 mutations are associated with mitochondrial dysfunction [10], and (iii) fibroblasts and some lymphocytes from SCBMS patients present mitochondrial alterations [10], it has been proposed that these additional symptoms are due to a defect in the mechanism of T cell activation [10].
A few cases of intellectual disability denominated mental retardation, autosomal recessive 47 (MRT47, MIM: 616193), are produced by mutations of FMN2 [38][39][41][42] (Figure 2, Supplementary Table S7). The genomic alterations consist of homozygous frameshift and nonsense mutations that are always found in consanguineous families, and large de novo heterozygous deletions. One case with sensory processing dysfunction was also associated with a de novo missense mutation [40]. This phenotype is consistent with the role of FMN2 in stabilizing filopodia tip adhesions and regulating the chemotaxis of neuronal grown cones [142][143]. Unlike the effect of FMN2 mutations in humans, Fmn2 KO mice do not present any alteration in the brain [144]. However, double-KO mice of FMN2 and filamin A show greater microcephaly severity and less neural progenitor proliferation compared with the phenotype of single filamin A KO mice. It has been suggested that this additive effect is a consequence of FMN2 and filamin A both forming part of the machinery of the endocytic route of the canonical Wnt pathway that regulates neural progenitor proliferation [145].
Mutations of other formin genes in addition to DIAPH1 and FMN2 have been associated with intellectual disability. In the case of INF2, some patients with FSGS and associated CMT, probably with severe mutations or an unfavorable genetic background, present intellectual disability and central nervous system anomalies [48][49][51]. An almost complete deletion of the FMNL2 gene has been associated with a case of mental retardation [146]. However, since the patient also presented haploinsufficiency in NR4A2, a gene involved in the cerebral dopaminergic system, it is difficult to ascertain whether the disorder is caused by one or both mutations.
2.5. Primary Ovarian Insufficiency
DIAPH2 has been implicated in premature ovarian failure (POF2A, MIM: 300511), also known as primary ovarian insufficiency, which manifests as premature menopause [29][30][31][32][33][34]. The patients generally present translocations of the X chromosome region that includes DIAPH2 (Figure 2, Supplementary Table S4). This gene might be involved in the development of gonads since it is expressed in the ovaries and testes of mouse embryos [29], and, indeed, some of the patients present ovarian dysgenesis (abnormal development) [33][34]. Underlining the importance of DIAPH2, Drosophila with mutations in Diaphanous are sterile due to cytokinesis failure that affects spermatogenesis in males and follicle cell division in females [147].
Consistent with the possibility that formin genes other than DIAPH2 are related to POF, FMN2 has been associated with POF and infertility in mice and in human patients [148][149]. Female Fmn2 KO mice exhibit defects in spindle positioning during meiosis I [144], which explains their low fertility. It has been proposed that upregulated levels of FMNL2 in humans also have a role in female infertility and gynecological health since they promote adenomyosis, which is characterized by the ectopic growth of the endometrium in the uterine walls, which are formed by the myometrium [150].
2.6. Cardiomyopathy
Thirteen out of the fifteen formins are expressed during postnatal development of the heart in mice within a specific timeframe that suggests a role for each formin in this process [151]. FHOD3, which is mainly expressed in the heart and regulates actin assembly in cardiomyocytes [152] (Figure 3B), has been linked to cardiac pathologies [153]. FHOD3 mutations have been associated with hypertrophic (CMH28, MIM: 619402) [95][96][97][98][99][100] and dilated cardiomyopathies [154] (Figure 2, Supplementary Table S9), which are conditions in which the walls of the heart becomes thicker and stiff, and where the heart is enlarged, respectively. As a consequence of these alterations, blood is pumped less effectively. Two intronic variants of FHOD3 have also been related to hypertrophic cardiomyopathy development [155] and a conservative substitution (p.Val1151Ile) with a reduced risk of dilated cardiomyopathy [156]. Fhod3 KO mice present embryonic lethality due to defects in cardiogenesis and in neural tube closure [157], whereas conditional KO mice show that the FHOD3 protein is needed not only for prenatal and postnatal heart development, but also for its maintenance, since adult mice present cardiomegaly and mild impairment of cardiac function [158]. Transgenic mice expressing FHOD3 defective in actin binding have a similar phenotype to that of dilated cardiomyopathy patients [157]. It is likely that the specific domain affected by the mutation, as well as the individual genetic background, could determine the appearance of one or other pathology, although both appear to be inherited in an autosomal-dominant manner. Angiotensin II is an important factor causing blood pressure overload-induced cardiac hypertrophy [159]. In cultured rat cardiomyocytes, angiotensin II signaling regulates FHOD3 activation through phosphorylation of its C-terminal region by ROCK kinase, raising the possibility that pathogenic FHOD3 causes heart hypertrophy by this mechanism [160].
References
- Schönichen, A.; Geyer, M. Fifteen Formins for an Actin Filament: A Molecular View on the Regulation of Human Formins. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 152–163.
- Chesarone, M.A.; DuPage, A.G.; Goode, B.L. Unleashing Formins to Remodel the Actin and Microtubule Cytoskeletons. Nat. Rev. Mol. Cell Biol. 2010, 11, 62–74.
- Goode, B.L.; Eck, M.J. Mechanism and Function of Formins in the Control of Actin Assembly. Annu. Rev. Biochem. 2007, 76, 593–627.
- Bartolini, F.; Gundersen, G.G. Formins and Microtubules. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 164–173.
- Fernandez-Barrera, J.; Alonso, M.A. Coordination of Microtubule Acetylation and the Actin Cytoskeleton by Formins. Cell. Mol. Life Sci. 2018, 75, 3181–3191.
- Lynch, E.D.; Lee, M.K.; Morrow, J.E.; Welcsh, P.L.; León, P.E.; King, M.-C. Nonsyndromic Deafness DFNA1 Associated with Mutation of a Human Homolog of the Drosophila Gene Diaphanous. Science 1997, 278, 1315–1318.
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.Org: Online Mendelian Inheritance in Man (OMIM®), an Online Catalog of Human Genes and Genetic Disorders. Nucleic Acids Res. 2015, 43, D789–D798.
- Hamosh, A.; Amberger, J.S.; Bocchini, C.; Scott, A.F.; Rasmussen, S.A. Online Mendelian Inheritance in Man (OMIM®): Victor McKusick’s Magnum Opus. Am. J. Med. Genet. Part A 2021.
- Iwasa, Y.; Nishio, S.; Usami, S. Comprehensive Genetic Analysis of Japanese Autosomal Dominant Sensorineural Hearing Loss Patients. PLoS ONE 2016, 11, e0166781.
- Kaustio, M.; Nayebzadeh, N.; Hinttala, R.; Tapiainen, T.; Åström, P.; Mamia, K.; Pernaa, N.; Lehtonen, J.; Glumoff, V.; Rahikkala, E.; et al. Loss of DIAPH1 Causes SCBMS, Combined Immunodeficiency, and Mitochondrial Dysfunction. J. Allergy Clin. Immunol. 2021, 148, 599–611.
- Brozkova, D.S.; Marková, S.P.; Mészárosová, A.U.; Jenčík, J.; Čejnová, V.; Čada, Z.; Laštůvková, J.; Rašková, D.; Seeman, P. Spectrum and Frequencies of Non GJB2 Gene Mutations in Czech Patients with Early Non-Syndromic Hearing Loss Detected by Gene Panel NGS and Whole-Exome Sequencing. Clin. Genet. 2020, 98, 548–554.
- Kim, B.J.; Ueyama, T.; Miyoshi, T.; Lee, S.; Han, J.H.; Park, H.-R.; Kim, A.R.; Oh, J.; Kim, M.Y.; Kang, Y.S.; et al. Differential Disruption of Autoinhibition and Defect in Assembly of Cytoskeleton during Cell Division Decide the Fate of Human DIAPH1-Related Cytoskeletopathy. J. Med. Genet. 2019, 56, 818–827.
- Kang, T.-H.; Baek, J.-I.; Sagong, B.; Park, H.-J.; Park, C.I.; Lee, K.-Y.; Kim, U.-K. A Novel Missense Variant in the DIAPH1 Gene in a Korean Family with Autosomal Dominant Nonsyndromic Hearing Loss. Genes Genet. Syst. 2016, 91, 289–292.
- Ercan-Sencicek, A.G.; Jambi, S.; Franjic, D.; Nishimura, S.; Li, M.; El-Fishawy, P.; Morgan, T.M.; Sanders, S.J.; Bilguvar, K.; Suri, M.; et al. Homozygous Loss of DIAPH1 Is a Novel Cause of Microcephaly in Humans. Eur. J. Hum. Genet. 2015, 23, 165–172.
- Sommen, M.; Schrauwen, I.; Vandeweyer, G.; Boeckx, N.; Corneveaux, J.J.; van den Ende, J.; Boudewyns, A.; De Leenheer, E.; Janssens, S.; Claes, K.; et al. DNA Diagnostics of Hereditary Hearing Loss: A Targeted Resequencing Approach Combined with a Mutation Classification System. Hum. Mutat. 2016, 37, 812–819.
- Al-Maawali, A.; Barry, B.J.; Rajab, A.; El-Quessny, M.; Seman, A.; Coury, S.N.; Barkovich, A.J.; Yang, E.; Walsh, C.A.; Mochida, G.H.; et al. Novel Loss-of-Function Variants in DIAPH1 Associated with Syndromic Microcephaly, Blindness, and Early Onset Seizures. Am. J. Med. Genet. Part A 2016, 170, 435–440.
- Yavarna, T.; Al-Dewik, N.; Al-Mureikhi, M.; Ali, R.; Al-Mesaifri, F.; Mahmoud, L.; Shahbeck, N.; Lakhani, S.; AlMulla, M.; Nawaz, Z.; et al. High Diagnostic Yield of Clinical Exome Sequencing in Middle Eastern Patients with Mendelian Disorders. Hum. Genet. 2015, 134, 967–980.
- Wu, K.; Wang, H.; Guan, J.; Lan, L.; Zhao, C.; Zhang, M.; Wang, D.; Wang, Q. A Novel Variant in Diaphanous Homolog 1 (DIAPH1) as the Cause of Auditory Neuropathy in a Chinese Family. Int. J. Pediatr. Otorhinolaryngol. 2020, 133, 109947.
- Westbury, S.K.; Downes, K.; Burney, C.; Lozano, M.L.; Obaji, S.G.; Toh, C.H.; Sevivas, T.; Morgan, N.V.; Erber, W.N.; Kempster, C.; et al. Phenotype Description and Response to Thrombopoietin Receptor Agonist in DIAPH1-Related Disorder. Blood Adv. 2018, 2, 2341–2346.
- Shearer, A.E.; Black-Ziegelbein, E.A.; Hildebrand, M.S.; Eppsteiner, R.W.; Ravi, H.; Joshi, S.; Guiffre, A.C.; Sloan, C.M.; Happe, S.; Howard, S.D.; et al. Advancing Genetic Testing for Deafness with Genomic Technology. J. Med. Genet. 2013, 50, 627–634.
- Ueyama, T.; Ninoyu, Y.; Nishio, S.; Miyoshi, T.; Torii, H.; Nishimura, K.; Sugahara, K.; Sakata, H.; Thumkeo, D.; Sakaguchi, H.; et al. Constitutive Activation of DIA1 (DIAPH1) via C-terminal Truncation Causes Human Sensorineural Hearing Loss. EMBO Mol. Med. 2016, 8, 1310–1324.
- Neuhaus, C.; Lang-Roth, R.; Zimmermann, U.; Heller, R.; Eisenberger, T.; Weikert, M.; Markus, S.; Knipper, M.; Bolz, H.J. Extension of the Clinical and Molecular Phenotype of DIAPH1-Associated Autosomal Dominant Hearing Loss (DFNA1). Clin. Genet. 2017, 91, 892–901.
- Bastida, J.M.; Lozano, M.L.; Benito, R.; Janusz, K.; Palma-Barqueros, V.; Del Rey, M.; Hernández-Sánchez, J.M.; Riesco, S.; Bermejo, N.; González-García, H.; et al. Introducing High-Throughput Sequencing into Mainstream Genetic Diagnosis Practice in Inherited Platelet Disorders. Haematologica 2018, 103, 148–162.
- Ganaha, A.; Kaname, T.; Shinjou, A.; Chinen, Y.; Yanagi, K.; Higa, T.; Kondo, S.; Suzuki, M. Progressive Macrothrombocytopenia and Hearing Loss in a Large Family with DIAPH1 Related Disease. Am. J. Med. Genet. 2017, 173, 2826–2830.
- Karki, N.R.; Ajebo, G.; Savage, N.; Kutlar, A. DIAPH1 Mutation as a Novel Cause of Autosomal Dominant Macrothrombocytopenia and Hearing Loss. Acta Haematol. 2021, 144, 87–90.
- Rabbolini, D.; Connor, D.; Morel-Kopp, M.-C.; Donikian, D.; Kondo, M.; Chen, W.; Alessi, M.-C.; Stevenson, W.; Chen, V.; Joseph, J.; et al. An Integrated Approach to Inherited Platelet Disorders: Results from a Research Collaborative, the Sydney Platelet Group. Pathology 2020, 52, 243–255.
- Stritt, S.; Nurden, P.; Turro, E.; Greene, D.; Jansen, S.B.; Westbury, S.K.; Petersen, R.; Astle, W.J.; Marlin, S.; Bariana, T.K.; et al. A Gain-of-Function Variant in DIAPH1 Causes Dominant Macrothrombocytopenia and Hearing Loss. Blood 2016, 127, 2903–2914.
- Ji, H.; Lu, J.; Wang, J.; Li, H.; Lin, X. Combined Examination of Sequence and Copy Number Variations in Human Deafness Genes Improves Diagnosis for Cases of Genetic Deafness. BMC Ear Nose Throat Disord. 2014, 14, 9.
- Bione, S.; Sala, C.; Manzini, C.; Arrigo, G.; Zuffardi, O.; Banfi, S.; Borsani, G.; Jonveaux, P.; Philippe, C.; Zuccotti, M.; et al. A Human Homologue of the Drosophila Melanogaster Diaphanous Gene Is Disrupted in a Patient with Premature Ovarian Failure: Evidence for Conserved Function in Oogenesis and Implications for Human Sterility. Am. J. Hum. Genet. 1998, 62, 533–541.
- Sala, C.; Arrigo, G.; Torri, G.; Martinazzi, F.; Riva, P.; Larizza, L.; Philippe, C.; Jonveaux, P.; Sloan, F.; Labella, T.; et al. Eleven X Chromosome Breakpoints Associated with Premature Ovarian Failure (POF) Map to a 15-Mb YAC Contig Spanning Xq21. Genomics 1997, 40, 123–131.
- Marozzi, A.; Manfredini, E.; Tibiletti, M.; Furlan, D.; Villa, N.; Vegetti, W.; Crosignani, P.; Ginelli, E.; Meneveri, R.; Dalprà, L. Molecular Definition of Xq Common-Deleted Region in Patients Affected by Premature Ovarian Failure. Hum. Genet. 2000, 107, 304–311.
- Misceo, D.; Rødningen, O.K.; Barøy, T.; Sorte, H.; Mellembakken, J.R.; Strømme, P.; Fannemel, M.; Frengen, E. A Translocation between Xq21.33 and 22q13.33 Causes an Intragenic SHANK3 Deletion in a Woman with Phelan-McDermid Syndrome and Hypergonadotropic Hypogonadism. Am. J. Med. Genet. 2011, 155, 403–408.
- Genesio, R.; Mormile, A.; Licenziati, M.R.; De Brasi, D.; Leone, G.; Balzano, S.; Izzo, A.; Bonfiglio, F.; Conti, A.; Fioretti, G.; et al. Short Stature and Primary Ovarian Insufficiency Possibly Due to Chromosomal Position Effect in a Balanced X;1 Translocation. Mol. Cytogenet. 2015, 8, 50.
- Bestetti, I.; Castronovo, C.; Sironi, A.; Caslini, C.; Sala, C.; Rossetti, R.; Crippa, M.; Ferrari, I.; Pistocchi, A.; Toniolo, D.; et al. High-Resolution Array-CGH Analysis on 46, XX Patients Affected by Early Onset Primary Ovarian Insufficiency Discloses New Genes Involved in Ovarian Function. Hum. Reprod. 2019, 34, 574–583.
- Sánchez-Martínez, A.; Benito-Orejas, J.I.; Tellería-Orriols, J.J.; Alonso-Ramos, M.J. Autosomal Dominant Auditory Neuropathy and Variant DIAPH3 (c.-173C>T). Acta Otorrinolaringol. Esp. 2017, 68, 183–185.
- Schoen, C.J.; Emery, S.B.; Thorne, M.C.; Ammana, H.R.; Śliwerska, E.; Arnett, J.; Hortsch, M.; Hannan, F.; Burmeister, M.; Lesperance, M.M. Increased Activity of Diaphanous Homolog 3 (DIAPH3)/Diaphanous Causes Hearing Defects in Humans with Auditory Neuropathy and in Drosophila. Proc. Natl. Acad. Sci. USA 2010, 107, 13396–13401.
- Schneider, R.; Deutsch, K.; Hoeprich, G.J.; Marquez, J.; Hermle, T.; Braun, D.A.; Seltzsam, S.; Kitzler, T.M.; Mao, Y.; Buerger, F.; et al. DAAM2 Variants Cause Nephrotic Syndrome via Actin Dysregulation. Am. J. Hum. Genet. 2020, 107, 1113–1128.
- Law, R.; Dixon-Salazar, T.; Jerber, J.; Cai, N.; Abbasi, A.A.; Zaki, M.S.; Mittal, K.; Gabriel, S.B.; Rafiq, M.A.; Khan, V.; et al. Biallelic Truncating Mutations in FMN2, Encoding the Actin-Regulatory Protein Formin 2, Cause Nonsyndromic Autosomal-Recessive Intellectual Disability. Am. J. Hum. Genet. 2014, 95, 721–728.
- Gorukmez, O.; Gorukmez, O.; Ekici, A. A Novel Nonsense FMN2 Mutation in Nonsyndromic Autosomal Recessive Intellectual Disability Syndrome. Fetal Pediatr. Pathol. 2020, 39, 1–5.
- Marco, E.J.; Aitken, A.B.; Nair, V.P.; da Gente, G.; Gerdes, M.R.; Bologlu, L.; Thomas, S.; Sherr, E.H. Burden of de Novo Mutations and Inherited Rare Single Nucleotide Variants in Children with Sensory Processing Dysfunction. BMC Med. Genom. 2018, 11, 50.
- Perrone, M.D.; Rocca, M.S.; Bruno, I.; Faletra, F.; Pecile, V.; Gasparini, P. De Novo 911 kb Interstitial Deletion on Chromosome 1q43 in a Boy with Mental Retardation and Short Stature. Eur. J. Med. Genet. 2012, 55, 117–119.
- Almuqbil, M.; Hamdan, F.F.; Mathonnet, G.; Rosenblatt, B.; Srour, M. De Novo Deletion of FMN2 in a Girl with Mild Non-Syndromic Intellectual Disability. Eur. J. Med. Genet. 2013, 56, 686–688.
- Brown, E.J.; Schlöndorff, J.S.; Becker, D.J.; Tsukaguchi, H.; Uscinski, A.L.; Higgs, H.N.; Henderson, J.M.; Pollak, M.R. Mutations in the Formin Protein INF2 Cause Focal Segmental Glomerulosclerosis. Nat. Genet. 2010, 42, 72–76.
- Nagano, C.; Yamamura, T.; Horinouchi, T.; Aoto, Y.; Ishiko, S.; Sakakibara, N.; Shima, Y.; Nakanishi, K.; Nagase, H.; Iijima, K.; et al. Comprehensive Genetic Diagnosis of Japanese Patients with Severe Proteinuria. Sci. Rep. 2020, 10, 270.
- Varner, J.D.; Chryst-Stangl, M.; Esezobor, C.I.; Solarin, A.; Wu, G.; Lane, B.; Hall, G.; Abeyagunawardena, A.; Matory, A.; Hunley, T.E.; et al. Genetic Testing for Steroid-Resistant-Nephrotic Syndrome in an Outbred Population. Front. Pediatr. 2018, 6, 307.
- Miao, J.; Pinto, E.; Vairo, F.; Hogan, M.C.; Erickson, S.B.; El Ters, M.; Bentall, A.J.; Kukla, A.; Greene, E.L.; Hernandez, L.H.; et al. Identification of Genetic Causes of Focal Segmental Glomerulosclerosis Increases with Proper Patient Selection. Mayo Clin. Proc. 2021, 96, 2342–2353.
- Laššuthová, P.; Šafka Brožková, D.; Krůtová, M.; Neupauerová, J.; Haberlová, J.; Mazanec, R.; Dřímal, P.; Seeman, P. Improving Diagnosis of Inherited Peripheral Neuropathies through Gene Panel Analysis. Orphanet J. Rare Dis. 2016, 11, 118.
- Boyer, O.; Nevo, F.; Plaisier, E.; Funalot, B.; Gribouval, O.; Benoit, G.; Huynh Cong, E.; Arrondel, C.; Tête, M.-J.; Montjean, R.; et al. INF2 Mutations in Charcot-Marie-Tooth Disease with Glomerulopathy. N. Engl. J. Med. 2011, 365, 2377–2388.
- Roos, A.; Weis, J.; Korinthenberg, R.; Fehrenbach, H.; Häusler, M.; Züchner, S.; Mache, C.; Hubmann, H.; Auer-Grumbach, M.; Senderek, J. Inverted Formin 2-Related Charcot-Marie-Tooth Disease: Extension of the Mutational Spectrum and Pathological Findings in Schwann Cells and Axons. J. Peripher. Nerv. Syst. 2015, 20, 52–59.
- Toyota, K.; Ogino, D.; Hayashi, M.; Taki, M.; Saito, K.; Abe, A.; Hashimoto, T.; Umetsu, K.; Tsukaguchi, H.; Hayasaka, K. INF2 Mutations in Charcot-Marie-Tooth Disease Complicated with Focal Segmental Glomerulosclerosis. J. Peripher. Nerv. Syst. 2013, 18, 97–98.
- Mademan, I.; Deconinck, T.; Dinopoulos, A.; Voit, T.; Schara, U.; Devriendt, K.; Meijers, B.; Lerut, E.; De Jonghe, P.; Baets, J. De Novo INF2 Mutations Expand the Genetic Spectrum of Hereditary Neuropathy with Glomerulopathy. Neurology 2013, 81, 1953–1958.
- Barua, M.; Brown, E.J.; Charoonratana, V.T.; Genovese, G.; Sun, H.; Pollak, M.R. Mutations in the INF2 Gene Account for a Significant Proportion of Familial but Not Sporadic Focal and Segmental Glomerulosclerosis. Kidney Int. 2013, 83, 316–322.
- Snoek, R.; Nguyen, T.Q.; van der Zwaag, B.; van Zuilen, A.D.; Kruis, H.M.E.; van Gils-Verrij, L.A.; Goldschmeding, R.; Knoers, N.V.A.M.; Rookmaaker, M.B.; van Eerde, A.M. Importance of Genetic Diagnostics in Adult-Onset Focal Segmental Glomerulosclerosis. Nephron 2019, 142, 351–358.
- Riedhammer, K.M.; Braunisch, M.C.; Günthner, R.; Wagner, M.; Hemmer, C.; Strom, T.M.; Schmaderer, C.; Renders, L.; Tasic, V.; Gucev, Z.; et al. Exome Sequencing and Identification of Phenocopies in Patients with Clinically Presumed Hereditary Nephropathies. Am. J. Kidney Dis. 2020, 76, 460–470.
- Boyer, O.; Benoit, G.; Gribouval, O.; Nevo, F.; Tête, M.-J.; Dantal, J.; Gilbert-Dussardier, B.; Touchard, G.; Karras, A.; Presne, C.; et al. Mutations in INF2 Are a Major Cause of Autosomal Dominant Focal Segmental Glomerulosclerosis. J. Am. Soc. Nephrol. 2011, 22, 239–245.
- Park, E.; Lee, C.; Kim, N.K.D.; Ahn, Y.H.; Park, Y.S.; Lee, J.H.; Kim, S.H.; Cho, M.H.; Cho, H.; Yoo, K.H.; et al. Genetic Study in Korean Pediatric Patients with Steroid-Resistant Nephrotic Syndrome or Focal Segmental Glomerulosclerosis. J. Clin. Med. 2020, 9, 2013.
- Rodriguez, P.Q.; Lohkamp, B.; Celsi, G.; Mache, C.J.; Auer-Grumbach, M.; Wernerson, A.; Hamajima, N.; Tryggvason, K.; Patrakka, J. Novel INF2 Mutation p. L77P in a Family with Glomerulopathy and Charcot-Marie-Tooth Neuropathy. Pediatr. Nephrol. 2013, 28, 339–343.
- Xie, J.; Hao, X.; Azeloglu, E.U.; Ren, H.; Wang, Z.; Ma, J.; Liu, J.; Ma, X.; Wang, W.; Pan, X.; et al. Novel Mutations in the Inverted Formin 2 Gene of Chinese Families Contribute to Focal Segmental Glomerulosclerosis. Kidney Int. 2015, 88, 593–604.
- Echaniz-Laguna, A.; Latour, P. A Cryptic Splicing Mutation in the INF2 Gene Causing Charcot-Marie-Tooth Disease with Minimal Glomerular Dysfunction. J. Peripher. Nerv. Syst. 2019, 24, 120–124.
- Challis, R.C.; Ring, T.; Xu, Y.; Wong, E.K.S.; Flossmann, O.; Roberts, I.S.D.; Ahmed, S.; Wetherall, M.; Salkus, G.; Brocklebank, V.; et al. Thrombotic Microangiopathy in Inverted Formin 2-Mediated Renal Disease. J. Am. Soc. Nephrol. 2017, 28, 1084–1091.
- Bacquet, J.; Stojkovic, T.; Boyer, A.; Martini, N.; Audic, F.; Chabrol, B.; Salort-Campana, E.; Delmont, E.; Desvignes, J.-P.; Verschueren, A.; et al. Molecular Diagnosis of Inherited Peripheral Neuropathies by Targeted Next-Generation Sequencing: Molecular Spectrum Delineation. BMJ Open 2018, 8, e021632.
- Lemieux, G.; Neemeh, J.A. Charcot-Marie-Tooth Disease and Nephritis. Can. Med. Assoc. J. 1967, 97, 1193–1198.
- Yao, T.; Udwan, K.; John, R.; Rana, A.; Haghighi, A.; Xu, L.; Hack, S.; Reich, H.N.; Hladunewich, M.A.; Cattran, D.C.; et al. Integration of Genetic Testing and Pathology for the Diagnosis of Adults with FSGS. Clin. J. Am. Soc. Nephrol. 2019, 14, 213–223.
- Caridi, G.; Lugani, F.; Dagnino, M.; Gigante, M.; Iolascon, A.; Falco, M.; Graziano, C.; Benetti, E.; Dugo, M.; Del Prete, D.; et al. Novel INF2 Mutations in an Italian Cohort of Patients with Focal Segmental Glomerulosclerosis, Renal Failure and Charcot-Marie-Tooth Neuropathy. Nephrol. Dial. Transplant. 2014, 29 (Suppl. 4), iv80–iv86.
- Sadowski, C.E.; Lovric, S.; Ashraf, S.; Pabst, W.L.; Gee, H.Y.; Kohl, S.; Engelmann, S.; Vega-Warner, V.; Fang, H.; Halbritter, J.; et al. A Single-Gene Cause in 29.5% of Cases of Steroid-Resistant Nephrotic Syndrome. J. Am. Soc. Nephrol. 2015, 26, 1279–1289.
- Fu, J.; Ma, M.; Pang, M.; Yang, L.; Li, G.; Song, J.; Zhang, J. Analysis of a pedigree with autosomal dominant intermediate Charcot-Marie-Tooth disease type E and nephropathy. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2019, 36, 918–921.
- Jin, S.; Wang, W.; Wang, R.; Lv, H.; Zhang, W.; Wang, Z.; Jiao, J.; Yuan, Y. INF2 Mutations Associated with Dominant Inherited Intermediate Charcot-Marie-Tooth Neuropathy with Focal Segmental Glomerulosclerosis in Two Chinese Patients. Clin. Neuropathol. 2015, 34, 275–281.
- Park, H.J.; Kim, H.J.; Hong, Y.B.; Nam, S.H.; Chung, K.W.; Choi, B.-O. A Novel INF2 Mutation in a Korean Family with Autosomal Dominant Intermediate Charcot-Marie-Tooth Disease and Focal Segmental Glomerulosclerosis. J. Peripher. Nerv. Syst. 2014, 19, 175–179.
- Laurin, L.-P.; Lu, M.; Mottl, A.K.; Blyth, E.R.; Poulton, C.J.; Weck, K.E. Podocyte-Associated Gene Mutation Screening in a Heterogeneous Cohort of Patients with Sporadic Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant. 2014, 29, 2062–2069.
- Wang, M.; Chun, J.; Genovese, G.; Knob, A.U.; Benjamin, A.; Wilkins, M.S.; Friedman, D.J.; Appel, G.B.; Lifton, R.P.; Mane, S.; et al. Contributions of Rare Gene Variants to Familial and Sporadic FSGS. J. Am. Soc. Nephrol. 2019, 30, 1625–1640.
- Münch, J.; Grohmann, M.; Lindner, T.H.; Bergmann, C.; Halbritter, J. Diagnosing FSGS without Kidney Biopsy—A Novel INF2-Mutation in a Family with ESRD of Unknown Origin. BMC Med. Genet. 2016, 17, 73.
- Büscher, A.K.; Celebi, N.; Hoyer, P.F.; Klein, H.-G.; Weber, S.; Hoefele, J. Mutations in INF2 May Be Associated with Renal Histology other than Focal Segmental Glomerulosclerosis. Pediatr. Nephrol. 2018, 33, 433–437.
- Braunisch, M.C.; Riedhammer, K.M.; Herr, P.-M.; Draut, S.; Günthner, R.; Wagner, M.; Weidenbusch, M.; Lungu, A.; Alhaddad, B.; Renders, L.; et al. Identification of Disease-Causing Variants by Comprehensive Genetic Testing with Exome Sequencing in Adults with Suspicion of Hereditary FSGS. Eur. J. Hum. Genet. 2021, 29, 262–270.
- Rood, I.M.; Bongers, E.M.H.F.; Lugtenberg, D.; Klein, I.H.H.T.; Steenbergen, E.J.; Wetzels, J.F.M.; Deegens, J.K.J. Familial Focal Segmental Glomerulosclerosis: Mutation in Inverted Formin 2 Mimicking Alport Syndrome. Neth. J. Med. 2016, 74, 82–85.
- Gbadegesin, R.A.; Lavin, P.J.; Hall, G.; Bartkowiak, B.; Homstad, A.; Jiang, R.; Wu, G.; Byrd, A.; Lynn, K.; Wolfish, N.; et al. Inverted Formin 2 Mutations with Variable Expression in Patients with Sporadic and Hereditary Focal and Segmental Glomerulosclerosis. Kidney Int. 2012, 81, 94–99.
- Warejko, J.K.; Tan, W.; Daga, A.; Schapiro, D.; Lawson, J.A.; Shril, S.; Lovric, S.; Ashraf, S.; Rao, J.; Hermle, T.; et al. Whole Exome Sequencing of Patients with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2018, 13, 53–62.
- Tan, W.; Lovric, S.; Ashraf, S.; Rao, J.; Schapiro, D.; Airik, M.; Shril, S.; Gee, H.Y.; Baum, M.; Daouk, G.; et al. Analysis of 24 Genes Reveals a Monogenic Cause in 11.1% of Cases with Steroid-Resistant Nephrotic Syndrome at a Single Center. Pediatr. Nephrol. 2018, 33, 305–314.
- Ogino, D.; Hashimoto, T.; Hattori, M.; Sugawara, N.; Akioka, Y.; Tamiya, G.; Makino, S.; Toyota, K.; Mitsui, T.; Hayasaka, K. Analysis of the Genes Responsible for Steroid-Resistant Nephrotic Syndrome and/or Focal Segmental Glomerulosclerosis in Japanese Patients by Whole-Exome Sequencing Analysis. J. Hum. Genet. 2016, 61, 137–141.
- Shang, S.; Peng, F.; Wang, T.; Wu, X.; Li, P.; Li, Q.; Chen, X.M. Genotype-Phenotype Correlation and Prognostic Impact in Chinese Patients with Alport Syndrome. Mol. Genet. Genom. Med. 2019, 7, e00741.
- Gribouval, O.; Boyer, O.; Hummel, A.; Dantal, J.; Martinez, F.; Sberro-Soussan, R.; Etienne, I.; Chauveau, D.; Delahousse, M.; Lionet, A.; et al. Identification of Genetic Causes for Sporadic Steroid-Resistant Nephrotic Syndrome in Adults. Kidney Int. 2018, 94, 1013–1022.
- Singh, A.; Singh, A.; Mishra, O.P.; Prasad, R.; Narayan, G.; Batra, V.V.; Tabatabaeifar, M.; Schaefer, F. Molecular Study of Childhood Steroid-Resistant Nephrotic Syndrome: A Hospital-Based Study. J. Pediatr. Genet. 2021.
- Santín, S.; Bullich, G.; Tazón-Vega, B.; García-Maset, R.; Giménez, I.; Silva, I.; Ruíz, P.; Ballarín, J.; Torra, R.; Ars, E. Clinical Utility of Genetic Testing in Children and Adults with Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2011, 6, 1139–1148.
- Larsen, C.P.; Durfee, T.; Wilson, J.D.; Beggs, M.L. A Custom Targeted Next-Generation Sequencing Gene Panel for the Diagnosis of Genetic Nephropathies. Am. J. Kidney Dis. 2016, 67, 992–993.
- Zhao, W.; Ma, X.; Zhang, X.; Luo, D.; Zhang, J.; Li, M.; Ye, Z.; Peng, H. INF2 p.Arg214Cys Mutation in a Chinese Family with Rapidly Progressive Renal Failure and Follow-up of Renal Transplantation: Case Report and Literature Review. BMC Nephrol. 2021, 22, 51.
- Safarikova, M.; Stekrova, J.; Honsova, E.; Horinova, V.; Tesar, V.; Reiterova, J. Mutational Screening of Inverted Formin 2 in Adult-Onset Focal Segmental Glomerulosclerosis or Minimal Change Patients from the Czech Republic. BMC Med. Genet. 2018, 19, 147.
- Gast, C.; Pengelly, R.J.; Lyon, M.; Bunyan, D.J.; Seaby, E.G.; Graham, N.; Venkat-Raman, G.; Ennis, S. Collagen (COL4A) Mutations Are the Most Frequent Mutations Underlying Adult Focal Segmental Glomerulosclerosis. Nephrol. Dial. Transplant. 2016, 31, 961–970.
- Lipska, B.S.; Iatropoulos, P.; Maranta, R.; Caridi, G.; Ozaltin, F.; Anarat, A.; Balat, A.; Gellermann, J.; Trautmann, A.; Erdogan, O.; et al. Genetic Screening in Adolescents with Steroid-Resistant Nephrotic Syndrome. Kidney Int. 2013, 84, 206–213.
- Bullich, G.; Trujillano, D.; Santín, S.; Ossowski, S.; Mendizábal, S.; Fraga, G.; Madrid, Á.; Ariceta, G.; Ballarín, J.; Torra, R.; et al. Targeted Next-Generation Sequencing in Steroid-Resistant Nephrotic Syndrome: Mutations in Multiple Glomerular Genes May Influence Disease Severity. Eur. J. Hum. Genet. 2015, 23, 1192–1199.
- Lee, H.K.; Han, K.H.; Jung, Y.H.; Kang, H.G.; Moon, K.C.; Ha, I.S.; Choi, Y.; Cheong, H.I. Variable Renal Phenotype in a Family with an INF2 Mutation. Pediatr. Nephrol. 2011, 26, 73–76.
- Sanchez-Ares, M.; Garcia-Vidal, M.; Antucho, E.-E.; Julio, P.; Eduardo, V.-M.; Lens, X.M.; Garcia-Gonzalez, M.A. A Novel Mutation, Outside of the Candidate Region for Diagnosis, in the Inverted Formin 2 Gene Can Cause Focal Segmental Glomerulosclerosis. Kidney Int. 2013, 83, 153–159.
- Büscher, A.K.; Beck, B.B.; Melk, A.; Hoefele, J.; Kranz, B.; Bamborschke, D.; Baig, S.; Lange-Sperandio, B.; Jungraithmayr, T.; Weber, L.T.; et al. Rapid Response to Cyclosporin A and Favorable Renal Outcome in Nongenetic Versus Genetic Steroid-Resistant Nephrotic Syndrome. Clin. J. Am. Soc. Nephrol. 2016, 11, 245–253.
- Weber, S.; Büscher, A.K.; Hagmann, H.; Liebau, M.C.; Heberle, C.; Ludwig, M.; Rath, S.; Alberer, M.; Beissert, A.; Zenker, M.; et al. Dealing with the Incidental Finding of Secondary Variants by the Example of SRNS Patients Undergoing Targeted Next-Generation Sequencing. Pediatr. Nephrol. 2016, 31, 73–81.
- Dohrn, M.F.; Glöckle, N.; Mulahasanovic, L.; Heller, C.; Mohr, J.; Bauer, C.; Riesch, E.; Becker, A.; Battke, F.; Hörtnagel, K.; et al. Frequent Genes in Rare Diseases: Panel-Based next Generation Sequencing to Disclose Causal Mutations in Hereditary Neuropathies. J. Neurochem. 2017, 143, 507–522.
- Sinha, R.; Maiti, R.; Das, D.; Mandal, K. Steroid Resistant Nephrotic Syndrome with Clumsy Gait Associated with INF2 Mutation. Indian Pediatr. 2020, 57, 764.
- Wu, G.; Ruan, J.; Liu, J.; Zhang, C.; Kang, L.; Wang, J.; Zou, Y.; Song, L. Variant Spectrum of Formin Homology 2 Domain-Containing 3 Gene in Chinese Patients with Hypertrophic Cardiomyopathy. J. Am. Heart Assoc. 2021, 10, e018236.
- Huang, S.; Pu, T.; Wei, W.; Xu, R.; Wu, Y. Exome Sequencing Identifies a FHOD3 p.S527del Mutation in a Chinese Family with Hypertrophic Cardiomyopathy. J. Gene Med. 2020, 22, e3146.
- Ochoa, J.P.; Sabater-Molina, M.; García-Pinilla, J.M.; Mogensen, J.; Restrepo-Córdoba, A.; Palomino-Doza, J.; Villacorta, E.; Martinez-Moreno, M.; Ramos-Maqueda, J.; Zorio, E.; et al. Formin Homology 2 Domain Containing 3 (FHOD3) Is a Genetic Basis for Hypertrophic Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 72, 2457–2467.
- Semsarian, C.; Ingles, J.; Bagnall, R.D. Revisiting Genome Sequencing Data in Light of Novel Disease Gene Associations. J. Am. Coll. Cardiol. 2019, 73, 1365–1366.
- Ochoa, J.P.; Lopes, L.R.; Perez-Barbeito, M.; Cazón-Varela, L.; de la Torre-Carpente, M.M.; Sonicheva-Paterson, N.; Uña-Iglesias, D.D.; Quinn, E.; Kuzmina-Krutetskaya, S.; Garrote, J.A.; et al. Deletions of Specific Exons of FHOD3 Detected by Next-Generation Sequencing Are Associated with Hypertrophic Cardiomyopathy. Clin. Genet. 2020, 98, 86–90.
- Hayashi, T.; Tanimoto, K.; Hirayama-Yamada, K.; Tsuda, E.; Ayusawa, M.; Nunoda, S.; Hosaki, A.; Kimura, A. Genetic Background of Japanese Patients with Pediatric Hypertrophic and Restrictive Cardiomyopathy. J. Hum. Genet. 2018, 63, 989–996.
- DeWard, A.D.; Eisenmann, K.M.; Matheson, S.F.; Alberts, A.S. The Role of Formins in Human Disease. Biochim. Biophys. Acta BBA Mol. Cell Res. 2010, 1803, 226–233.
- Randall, T.S.; Ehler, E. A Formin-g Role during Development and Disease. Eur. J. Cell Biol. 2014, 93, 205–211.
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410.
- Scott, R.P.; Quaggin, S.E. The Cell Biology of Renal Filtration. J. Cell Biol. 2015, 209, 199–210.
- Rosenberg, A.Z.; Kopp, J.B. Focal Segmental Glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2017, 12, 502–517.
- Labat-de-Hoz, L.; Alonso, M.A. The Formin INF2 in Disease: Progress from 10 Years of Research. Cell. Mol. Life Sci. 2020, 77, 4581–4600.
- Bose, B.; Cattran, D. Glomerular Diseases: FSGS. Clin. J. Am. Soc. Nephrol. 2014, 9, 626–632.
- Fogo, A.B. Causes and Pathogenesis of Focal Segmental Glomerulosclerosis. Nat. Rev. Nephrol. 2015, 11, 76–87.
- Rossor, A.M.; Polke, J.M.; Houlden, H.; Reilly, M.M. Clinical Implications of Genetic Advances in Charcot-Marie-Tooth Disease. Nat. Rev. Neurol. 2013, 9, 562–571.
- Bayraktar, S.; Nehrig, J.; Menis, E.; Karli, K.; Janning, A.; Struk, T.; Halbritter, J.; Michgehl, U.; Krahn, M.P.; Schuberth, C.E.; et al. A Deregulated Stress Response Underlies Distinct INF2-Associated Disease Profiles. J. Am. Soc. Nephrol. 2020, 31, 1296–1313.
- Mu, A.; Fung, T.S.; Kettenbach, A.N.; Chakrabarti, R.; Higgs, H.N. A Complex Containing Lysine-Acetylated Actin Inhibits the Formin INF2. Nat. Cell Biol. 2019, 21, 592–602.
- Subramanian, B.; Sun, H.; Yan, P.; Charoonratana, V.T.; Higgs, H.N.; Wang, F.; Lai, K.-M.V.; Valenzuela, D.M.; Brown, E.J.; Schlöndorff, J.S.; et al. Mice with Mutant Inf2 Show Impaired Podocyte and Slit Diaphragm Integrity in Response to Protamine-Induced Kidney Injury. Kidney Int. 2016, 90, 363–372.
- Mathis, S.; Funalot, B.; Boyer, O.; Lacroix, C.; Marcorelles, P.; Magy, L.; Richard, L.; Antignac, C.; Vallat, J.-M. Neuropathologic Characterization of INF2-Related Charcot-Marie-Tooth Disease: Evidence for a Schwann Cell Actinopathy. J. Neuropathol. Exp. Neurol. 2014, 73, 223–233.
- Tricaud, N. Myelinating Schwann Cell Polarity and Mechanically-Driven Myelin Sheath Elongation. Front. Cell. Neurosci. 2018, 11, 414.
- Takemon, Y.; Wright, V.; Davenport, B.; Gatti, D.M.; Sheehan, S.M.; Letson, K.; Savage, H.S.; Lennon, R.; Korstanje, R. Uncovering Modifier Genes of X-Linked Alport Syndrome Using a Novel Multiparent Mouse Model. J. Am. Soc. Nephrol. 2021, 32, 1961–1973.
- Schwander, M.; Kachar, B.; Müller, U. Review Series: The Cell Biology of Hearing. J. Cell Biol. 2010, 190, 9–20.
- Drummond, M.C.; Belyantseva, I.A.; Friderici, K.H.; Friedman, T.B. Actin in Hair Cells and Hearing Loss. Hear Res. 2012, 288, 89–99.
- Wallar, B.J.; Stropich, B.N.; Schoenherr, J.A.; Holman, H.A.; Kitchen, S.M.; Alberts, A.S. The Basic Region of the Diaphanous-Autoregulatory Domain (DAD) Is Required for Autoregulatory Interactions with the Diaphanous-Related Formin Inhibitory Domain. J. Biol. Chem. 2006, 281, 4300–4307.
- Miyoshi, T.; Belyantseva, I.A.; Kitajiri, S.-I.; Miyajima, H.; Nishio, S.-Y.; Usami, S.-I.; Kim, B.J.; Choi, B.Y.; Omori, K.; Shroff, H.; et al. Human Deafness-Associated Variants Alter the Dynamics of Key Molecules in Hair Cell Stereocilia F-Actin Cores. Hum. Genet. 2021.
- Lakha, R.; Montero, A.M.; Jabeen, T.; Costeas, C.C.; Ma, J.; Vizcarra, C.L. Variable Autoinhibition among Deafness-Associated Variants of Diaphanous 1 (DIAPH1). Biochemistry 2021, 60, 2320–2329.
- Ninoyu, Y.; Sakaguchi, H.; Lin, C.; Suzuki, T.; Hirano, S.; Hisa, Y.; Saito, N.; Ueyama, T. The Integrity of Cochlear Hair Cells Is Established and Maintained through the Localization of Dia1 at Apical Junctional Complexes and Stereocilia. Cell Death Dis. 2020, 11, 536.
- Schoen, C.J.; Burmeister, M.; Lesperance, M.M. Diaphanous Homolog 3 (Diap3) Overexpression Causes Progressive Hearing Loss and Inner Hair Cell Defects in a Transgenic Mouse Model of Human Deafness. PLoS ONE 2013, 8, e56520.
- Surel, C.; Guillet, M.; Lenoir, M.; Bourien, J.; Sendin, G.; Joly, W.; Delprat, B.; Lesperance, M.M.; Puel, J.-L.; Nouvian, R. Remodeling of the Inner Hair Cell Microtubule Meshwork in a Mouse Model of Auditory Neuropathy AUNA1. eNeuro 2016, 3, 0295-16.2016.
- Bae, S.-H.; Baek, J.-I.; Lee, J.D.; Song, M.H.; Kwon, T.-J.; Oh, S.-K.; Jeong, J.Y.; Choi, J.Y.; Lee, K.-Y.; Kim, U.-K. Genetic Analysis of Auditory Neuropathy Spectrum Disorder in the Korean Population. Gene 2013, 522, 65–69.
- Almazni, I.; Stapley, R.; Morgan, N.V. Inherited Thrombocytopenia: Update on Genes and Genetic Variants which May Be Associated with Bleeding. Front. Cardiovasc. Med. 2019, 6, 80.
- Becker, I.C.; Scheller, I.; Wackerbarth, L.M.; Beck, S.; Heib, T.; Aurbach, K.; Manukjan, G.; Gross, C.; Spindler, M.; Nagy, Z.; et al. Actin/Microtubule Crosstalk during Platelet Biogenesis in Mice Is Critically Regulated by Twinfilin1 and Cofilin1. Blood Adv. 2020, 4, 2124–2134.
- Italiano, J.E.; Lecine, P.; Shivdasani, R.A.; Hartwig, J.H. Blood Platelets Are Assembled Principally at the Ends of Proplatelet Processes Produced by Differentiated Megakaryocytes. J. Cell Biol. 1999, 147, 1299–1312.
- Patel, S.R. The Biogenesis of Platelets from Megakaryocyte Proplatelets. J. Clin. Investig. 2005, 115, 3348–3354.
- Potts, K.S.; Farley, A.; Dawson, C.A.; Rimes, J.; Biben, C.; de Graaf, C.; Potts, M.A.; Stonehouse, O.J.; Carmagnac, A.; Gangatirkar, P.; et al. Membrane Budding Is a Major Mechanism of in Vivo Platelet Biogenesis. J. Exp. Med. 2020, 217, e20191206.
- Pan, J.; Lordier, L.; Meyran, D.; Rameau, P.; Lecluse, Y.; Kitchen-Goosen, S.; Badirou, I.; Mokrani, H.; Narumiya, S.; Alberts, A.S.; et al. The Formin DIAPH1 (MDia1) Regulates Megakaryocyte Proplatelet Formation by Remodeling the Actin and Microtubule Cytoskeletons. Blood 2014, 124, 3967–3977.
- Zuidscherwoude, M.; Green, H.L.H.; Thomas, S.G. Formin Proteins in Megakaryocytes and Platelets: Regulation of Actin and Microtubule Dynamics. Platelets 2019, 30, 23–30.
- Ishizaki, T.; Morishima, Y.; Okamoto, M.; Furuyashiki, T.; Kato, T.; Narumiya, S. Coordination of Microtubules and the Actin Cytoskeleton by the Rho Effector MDia1. Nat. Cell Biol. 2001, 3, 8–14.
- Palazzo, A.F.; Cook, T.A.; Alberts, A.S.; Gundersen, G.G. mDia Mediates Rho-Regulated Formation and Orientation of Stable Microtubules. Nat. Cell Biol. 2001, 3, 723–729.
- Heremans, J.; Garcia-Perez, J.E.; Turro, E.; Schlenner, S.M.; Casteels, I.; Collin, R.; de Zegher, F.; Greene, D.; Humblet-Baron, S.; Lesage, S.; et al. Abnormal Differentiation of B Cells and Megakaryocytes in Patients with Roifman Syndrome. J. Allergy Clin. Immunol. 2018, 142, 630–646.
- Merico, D.; Roifman, M.; Braunschweig, U.; Yuen, R.K.C.; Alexandrova, R.; Bates, A.; Reid, B.; Nalpathamkalam, T.; Wang, Z.; Thiruvahindrapuram, B.; et al. Compound Heterozygous Mutations in the Noncoding RNU4ATAC Cause Roifman Syndrome by Disrupting Minor Intron Splicing. Nat. Commun. 2015, 6, 8718.
- Dutta, P.; Maiti, S. Expression of Multiple Formins in Adult Tissues and during Developmental Stages of Mouse Brain. Gene Expr. Patterns 2015, 19, 52–59.
- Kawabata Galbraith, K.; Kengaku, M. Multiple Roles of the Actin and Microtubule-Regulating Formins in the Developing Brain. Neurosci. Res. 2019, 138, 59–69.
- Damiani, D.; Goffinet, A.M.; Alberts, A.; Tissir, F. Lack of Diaph3 Relaxes the Spindle Checkpoint Causing the Loss of Neural Progenitors. Nat. Commun. 2016, 7, 13509.
- Thumkeo, D.; Shinohara, R.; Watanabe, K.; Takebayashi, H.; Toyoda, Y.; Tohyama, K.; Ishizaki, T.; Furuyashiki, T.; Narumiya, S. Deficiency of mDia, an Actin Nucleator, Disrupts Integrity of Neuroepithelium and Causes Periventricular Dysplasia. PLoS ONE 2011, 6, e25465.
- Eisenmann, K.M.; West, R.A.; Hildebrand, D.; Kitchen, S.M.; Peng, J.; Sigler, R.; Zhang, J.; Siminovitch, K.A.; Alberts, A.S. T Cell Responses in Mammalian Diaphanous-Related Formin MDia1 Knock-out Mice. J. Biol. Chem. 2007, 282, 25152–25158.
- Sakata, D.; Taniguchi, H.; Yasuda, S.; Adachi-Morishima, A.; Hamazaki, Y.; Nakayama, R.; Miki, T.; Minato, N.; Narumiya, S. Impaired T Lymphocyte Trafficking in Mice Deficient in an Actin-Nucleating Protein, MDia1. J. Exp. Med. 2007, 204, 2031–2038.
- Kundu, T.; Dutta, P.; Nagar, D.; Maiti, S.; Ghose, A. Coupling of Dynamic Microtubules to F-Actin by Fmn2 Regulates Chemotaxis of Neuronal Growth Cones. J. Cell Sci. 2021, 134, jcs252916.
- Sahasrabudhe, A.; Ghate, K.; Mutalik, S.; Jacob, A.; Ghose, A. Formin 2 Regulates the Stabilization of Filopodial Tip Adhesions in Growth Cones and Affects Neuronal Outgrowth and Pathfinding in Vivo. Development 2016, 143, 449–460.
- Leader, B.; Lim, H.; Carabatsos, M.J.; Harrington, A.; Ecsedy, J.; Pellman, D.; Maas, R.; Leder, P. Formin-2, Polyploidy, Hypofertility and Positioning of the Meiotic Spindle in Mouse Oocytes. Nat. Cell Biol. 2002, 4, 921–928.
- Lian, G.; Dettenhofer, M.; Lu, J.; Downing, M.; Chenn, A.; Wong, T.; Sheen, V. Filamin A- and Formin 2-Dependent Endocytosis Regulates Proliferation via the Canonical Wnt Pathway. Development 2016, 143, 4509–4520.
- Lybaek, H.; Ørstavik, K.H.; Prescott, T.; Hovland, R.; Breilid, H.; Stansberg, C.; Steen, V.M.; Houge, G. An 8.9 Mb 19p13 Duplication Associated with Precocious Puberty and a Sporadic 3.9 Mb 2q23.3q24.1 Deletion Containing NR4A2 in Mentally Retarded Members of a Family with an Intrachromosomal 19p-into-19q between-Arm Insertion. Eur. J. Hum. Genet. 2009, 17, 904–910.
- Castrillon, D.H.; Wasserman, S.A. Diaphanous Is Required for Cytokinesis in Drosophila and Shares Domains of Similarity with the Products of the Limb Deformity Gene. Development 1994, 120, 3367–3377.
- Ryley, D.A.; Wu, H.-H.; Leader, B.; Zimon, A.; Reindollar, R.H.; Gray, M.R. Characterization and Mutation Analysis of the Human Formin-2 (FMN2) Gene in Women with Unexplained Infertility. Fertil. Steril. 2005, 83, 1363–1371.
- Tšuiko, O.; Nõukas, M.; Žilina, O.; Hensen, K.; Tapanainen, J.S.; Mägi, R.; Kals, M.; Kivistik, P.A.; Haller-Kikkatalo, K.; Salumets, A.; et al. Copy Number Variation Analysis Detects Novel Candidate Genes Involved in Follicular Growth and Oocyte Maturation in a Cohort of Premature Ovarian Failure Cases. Hum. Reprod. 2016, 31, 1913–1925.
- Li, Y.; Zhang, Q.; Liu, F.; Zhang, Z.; Zou, Y.; Yang, B.; Luo, Y.; Wang, L.; Huang, O. Inhibition of Formin like 2 Promotes the Transition of Ectopic Endometrial Stromal Cells to Epithelial Cells in Adenomyosis through a MET-like Process. Gene 2019, 710, 186–192.
- Rosado, M.; Barber, C.F.; Berciu, C.; Feldman, S.; Birren, S.J.; Nicastro, D.; Goode, B.L. Critical Roles for Multiple Formins during Cardiac Myofibril Development and Repair. Mol. Biol. Cell 2014, 25, 811–827.
- Taniguchi, K.; Takeya, R.; Suetsugu, S.; Kan-o, M.; Narusawa, M.; Shiose, A.; Tominaga, R.; Sumimoto, H. Mammalian Formin Fhod3 Regulates Actin Assembly and Sarcomere Organization in Striated Muscles. J. Biol. Chem. 2009, 284, 29873–29881.
- Ehler, E. Actin-Associated Proteins and Cardiomyopathy-the “unknown” beyond Troponin and Tropomyosin. Biophys. Rev. 2018, 10, 1121–1128.
- Arimura, T.; Takeya, R.; Ishikawa, T.; Yamano, T.; Matsuo, A.; Tatsumi, T.; Nomura, T.; Sumimoto, H.; Kimura, A. Dilated Cardiomyopathy-Associated FHOD3 Variant Impairs the Ability to Induce Activation of Transcription Factor Serum Response Factor. Circ. J. 2013, 77, 2990–2996.
- Harper, A.R.; Goel, A.; Grace, C.; Thomson, K.L.; Petersen, S.E.; Xu, X.; Waring, A.; Ormondroyd, E.; Kramer, C.M.; Ho, C.Y.; et al. Common Genetic Variants and Modifiable Risk Factors Underpin Hypertrophic Cardiomyopathy Susceptibility and Expressivity. Nat. Genet. 2021, 53, 135–142.
- Esslinger, U.; Garnier, S.; Korniat, A.; Proust, C.; Kararigas, G.; Müller-Nurasyid, M.; Empana, J.-P.; Morley, M.P.; Perret, C.; Stark, K.; et al. Exome-Wide Association Study Reveals Novel Susceptibility Genes to Sporadic Dilated Cardiomyopathy. PLoS ONE 2017, 12, e0172995.
- Kan, O.M.; Takeya, R.; Abe, T.; Kitajima, N.; Nishida, M.; Tominaga, R.; Kurose, H.; Sumimoto, H. Mammalian Formin Fhod3 Plays an Essential Role in Cardiogenesis by Organizing Myofibrillogenesis. Biol. Open 2012, 1, 889–896.
- Ushijima, T.; Fujimoto, N.; Matsuyama, S.; Kan, O.M.; Kiyonari, H.; Shioi, G.; Kage, Y.; Yamasaki, S.; Takeya, R.; Sumimoto, H. The Actin-Organizing Formin Protein Fhod3 Is Required for Postnatal Development and Functional Maintenance of the Adult Heart in Mice. J. Biol. Chem. 2018, 293, 148–162.
- Heineke, J.; Molkentin, J.D. Regulation of Cardiac Hypertrophy by Intracellular Signalling Pathways. Nat. Rev. Mol. Cell Biol. 2006, 7, 589–600.
- Zhou, Q.; Wei, S.-S.; Wang, H.; Wang, Q.; Li, W.; Li, G.; Hou, J.-W.; Chen, X.-M.; Chen, J.; Xu, W.-P.; et al. Crucial Role of ROCK2-Mediated Phosphorylation and Upregulation of FHOD3 in the Pathogenesis of Angiotensin II-Induced Cardiac Hypertrophy. Hypertension 2017, 69, 1070–1083.
More
Information
Subjects:
Genetics & Heredity
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
18 Oct 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No