 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Cappuccilli | + 3648 word(s) | 3648 | 2021-10-14 11:17:44 | | | |
| 2 | Peter Tang | Meta information modification | 3648 | 2021-10-15 08:57:25 | | |
Video Upload Options
Patients affected by chronic kidney disease (CKD) or end-stage renal disease (ESRD) experience a huge cardiovascular risk and cardiovascular events represent the leading causes of death. Folic acid and vitamin B12 could not only be mere cofactors in the homocysteine metabolism; they may have a direct action in determining tissue damage and cardiovascular risk.
1. Introduction
2. B Vitamins—Homocysteine Pathway
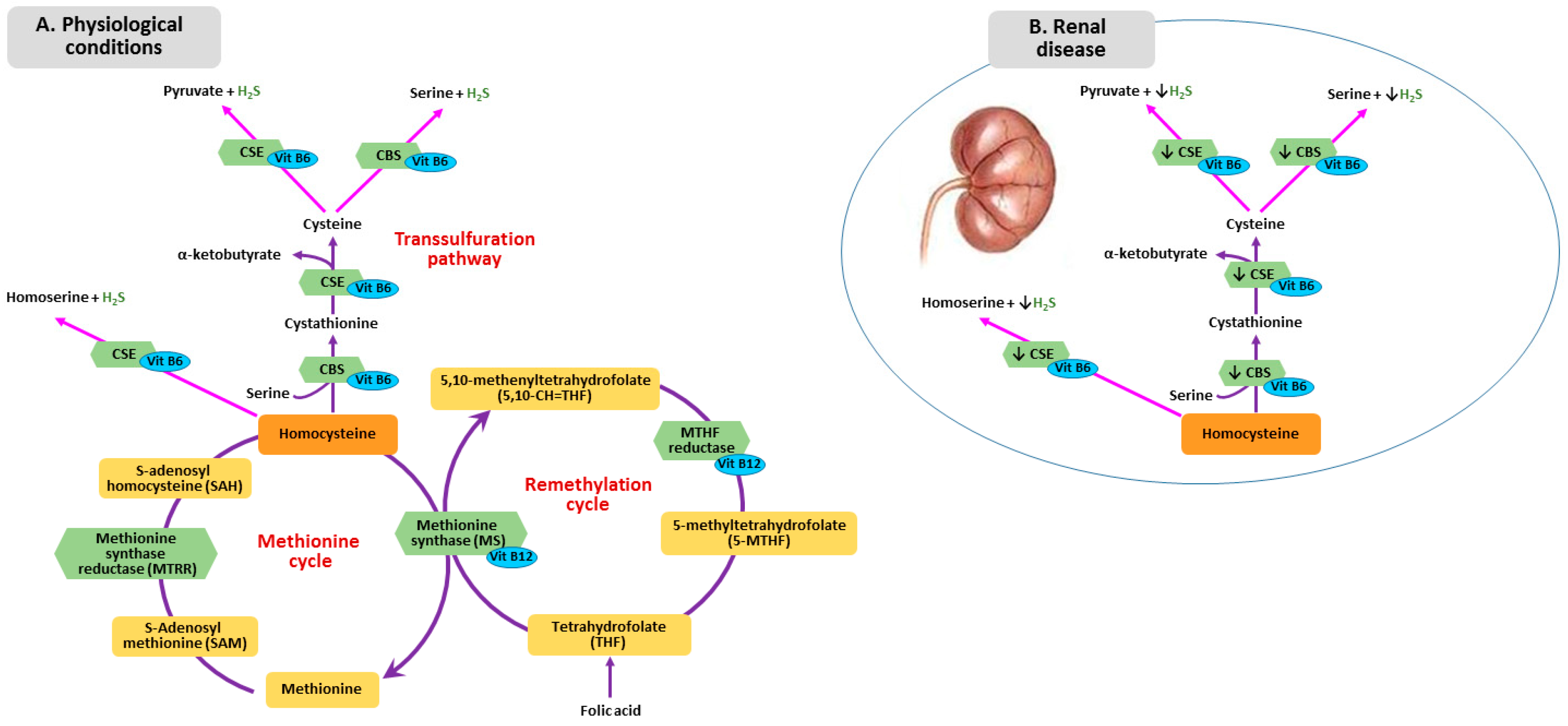
3. Metabolism of Homocysteine, Folic Acid and Vitamin B12 in CKD
4. Homocysteine-Mediated Tissue Damage
5. Folic Acid and Vitamin B12 Impairment and Tissue Injury
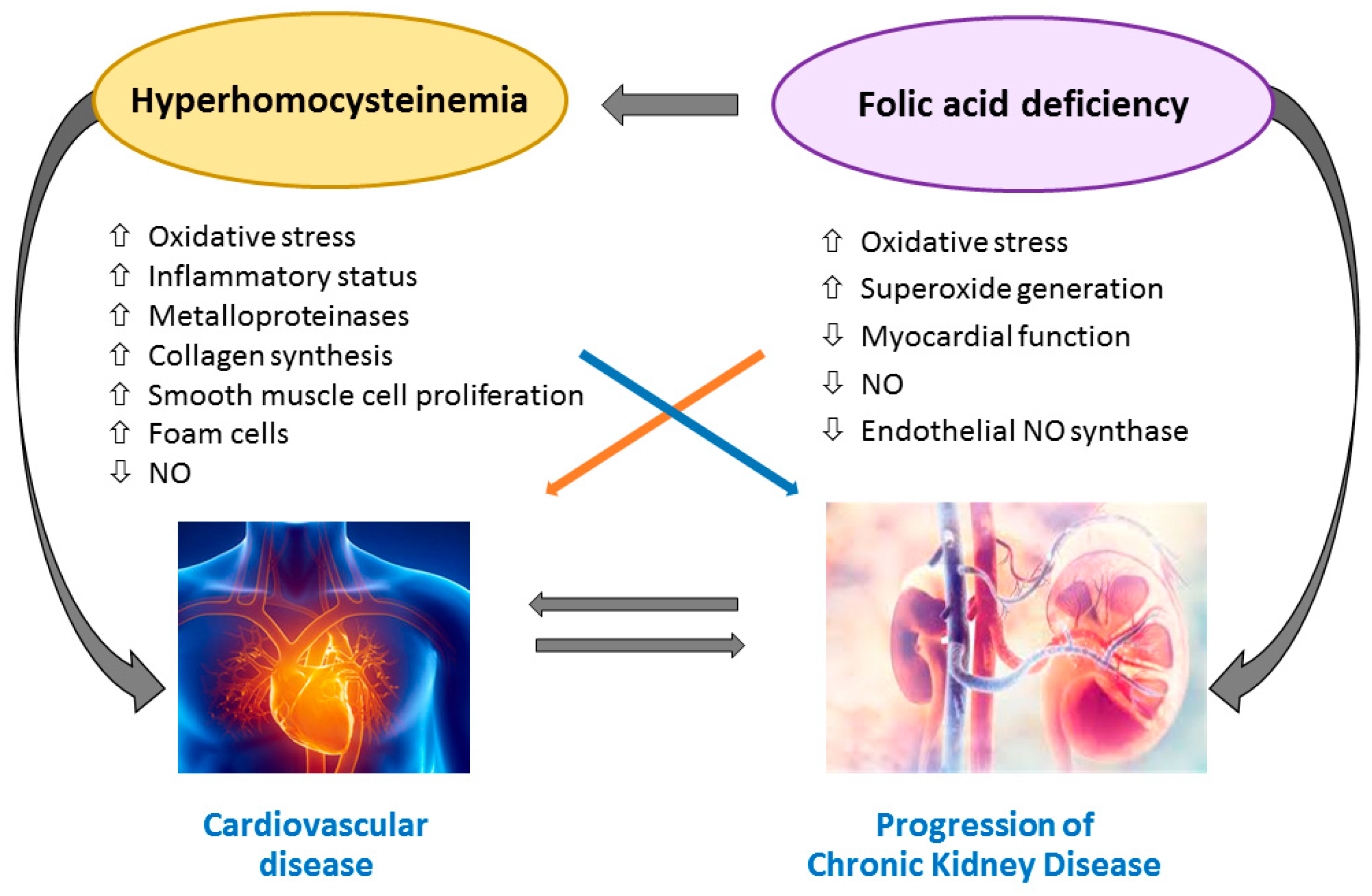
6. Effect of Folic Acid and Vitamin B12 Supplementation on CVD and Mortality in CKD and ESRD
|
Study |
Design/Intervention |
Participants, n |
End Point |
Follow-up, Years |
Results |
|---|---|---|---|---|---|
|
Xu et al., 2016 [79] |
Double blind RCT: enalapril 10 mg versus enalapril 10 mg plus folic acid |
15,104 (eGFR ≥ 30 mL/min). No folic acid fortification |
CKD progression |
4.4 |
Enalapril plus folic acid delayed CKD progression |
|
House et al., 2010 [80] |
Double blind RCT: folic acid 2.5 mg + Vitamin B6 25 mg + Vitamin B12 1 mg versus placebo |
238 (diabetic nephropathy with eGFR > 30 mL/min). Folic acid fortification |
CKD progression |
2.6 |
Greater GFR decrease and more CVD events in treatment group |
|
Heinz et al., 2010 [77] |
Double blind RCT: folic acid 5 mg, vitamin B12 50 µg, vitamin B6 20 mg versus placebo 3 times a week |
650 hemodialysis patients |
All-cause mortality, cardiovascular events |
2 |
No differences |
|
Mann et al., 2008 [81] |
Double blind RCT: folic acid 2.5 mg + vitamin B6 50 mg + vitamin B12 1 mg versus placebo |
619 CKD (eGFR <60 mL/min) |
All-cause mortality, cardiovascular events |
5 |
No differences |
|
Cianciolo et al., 2008 [11] |
Open label randomized trial: 5-MTHF intravenous. three times a week versus folic acid 5 mg oral daily |
314 hemodialysis patients |
All-cause mortality |
4.5 |
Less mortality risk in 5-MTHF group (independent of homocysteine) |
|
Jamison et al., 2007 [76] |
Double blind RCT (HOST): folic acid 40 mg + vitamin B6 100 mg + vitamin B12 2 mg versus placebo |
2056 CKD (eGFR ≤ 30) or hemodialysis (folic acid fortification) |
All-cause mortality, CKD progression |
3.2 |
No differences |
|
Vianna et al., 2007 [82] |
Double blind RCT: folic acid 5 mg versus placebo |
97 hemodialysis patients |
Cardiovascular events |
2 |
No differences |
|
Zoungas et al., 2006 [75] |
Double blind RCT (ASFAST): folic acid 15 mg versus placebo |
315 CKD (eGFR < 25 mL/min), hemodialysis and peritoneal dialysis |
Cardiovascular events and mortality |
3.6 |
No differences |
|
Righetti et al., 2006 [83] |
Open prospective trial: folic acid 5 mg versus untreated |
114 hemodialysis patients |
Cardiovascular events |
2.4 |
Folic acid decreases CVD events |
|
Wrone et al., 2004 [74] |
Three arms, double blind RCT: folic acid 1 mg or 5 mg or 15 mg |
510 hemodialysis patients |
Cardiovascular events and mortality |
2 |
No differences |
|
Righetti et al., 2003 [73] |
Placebo-controlled, non-blinded RCT: folic acid 5, 15, 25 mg or placebo |
81 hemodialysis patients |
Cardiovascular mortality |
1 |
No differences |
Abbreviations: CKD, Chronic Kidney Disease; CVD, Cardiovascular Disease; eGFR, estimated Glomerular Filtration Rate; RCT, Randomized Clinical Trial.
References
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305.
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S16–S23.
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272.
- McCullough, P.A.; Steigerwalt, S.; Tolia, K.; Chen, S.C.; Li, S.; Norris, K.C.; Whaley-Connell, A.; KEEP Investigators. Cardiovascular disease in chronic kidney disease: Data from the Kidney Early Evaluation Program (KEEP). Curr. Diab. Rep. 2011, 11, 47–55.
- Ekdahl, K.; Soveri, I.; Hilborn, J.; Fellström, B.; Nilsson, B. Cardiovascular disease in hemodialysis: Role of the intravascular innate immune system. Nat. Rev. Nephrol. 2017, 13, 285–296.
- McCully, K.S. Homocysteine and vascular disease. Nat. Med. 1996, 2, 386–389.
- Chrysant, S.G.; Chrysant, G.S. The current status of homocysteine as a risk factor for cardiovascular disease: A mini review. Expert. Rev. Cardiovasc. Ther. 2018, 16, 559–565.
- Robinson, K. Renal disease, homocysteine, and cardiovascular complications. Circulation 2004, 109, 294–295.
- Suliman, M.E.; Lindholm, B.; Barany, P.; Qureshi, A.R.; Stenvinkel, P. Homocysteine-lowering is not a primary target for cardiovascular disease prevention in chronic kidney disease patients. Semin. Dial. 2007, 20, 523–529.
- Heinz, J.; Kropf, S.; Luley, C.; Dierkes, J. Homocysteine as a risk factor for cardiovascular disease in patients treated by dialysis: A meta-analysis. Am. J. Kidney Dis. 2009, 54, 478–489.
- Cianciolo, G.; La Manna, G.; Colì, L.; Donati, G.; D’Addio, F.; Persici, E.; Comai, G.; Wratten, M.; Dormi, A.; Mantovani, V.; et al. 5-methyltetrahydrofolate administration is associated with prolonged survival and reduced inflammation in ESRD patients. Am. J. Nephrol. 2008, 28, 941–948.
- Cianciolo, G.; De Pascalis, A.; Di Lullo, L.; Ronco, C.; Zannini, C.; La Manna, G. Folic Acid and Homocysteine in Chronic Kidney Disease and Cardiovascular Disease Progression: Which Comes First? Cardiorenal. Med. 2017, 7, 255–266.
- Marti, F.; Vollenweider, P.; Marques-Vidal, P.M.; Mooser, V.; Waeber, G.; Paccaud, F.; Bochud, M. Hyperhomocysteinemia is independently associated with albuminuria in the population-based CoLaus study. BMC Public Health. 2011, 11.
- Ponte, B.; Pruijm, M.; Marques-Vidal, P.; Martin, P.Y.; Burnier, M.; Paccaud, F.; Waeber, G.; Vollenweider, P.; Bochud, M. Determinants and burden of chronic kidney disease in the population-based CoLaus study: A cross-sectional analysis. Nephrol. Dial. Transpl. 2013, 28, 2329–2339.
- Soohoo, M.; Ahmadi, S.F.; Qader, H.; Streja, E.; Obi, Y.; Moradi, H.; Rhee, C.M.; Kim, T.H.; Kovesdy, C.P.; Kalantar-Zadeh, K. Association of serum vitamin B12 and folate with mortality in incident hemodialysis patients. Nephrol. Dial. Transpl. 2017, 32, 1024–1032.
- Kang, S.S.; Wong, P.W.K.; Malinow, M.R. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu. Rev. Nutr. 1992, 12, 279–298.
- Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Vitamin B12: Unique metalorganic compounds and the most complex vitamins. Molecules 2010, 15, 3228–3259.
- Long, Y.; Nie, J. Homocysteine in Renal Injury. Kidney Dis. 2016, 2, 80–87.
- Van Guldener, C.; Stam, F.; Stehouwer, C.D. Hyperhomocysteinaemia in chronic kidney disease: Focus on transmethylation. Clin. Chem. Lab. Med. 2005, 43, 1026–1031.
- Jacobsen, D.W. Homocysteine and vitamins in cardiovascular disease. Clin. Chem. 1998, 44, 1833–1843.
- Cianciolo, G.; Cappuccilli, M.; La Manna, G. The Hydrogen Sulfide-Vitamin B12-Folic Acid Axis: An Intriguing Issue in Chronic Kidney Disease. A Comment on Toohey JI: “Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer”. Molecules 2017, 22, 1216.
- Toohey, J.I. Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer. Molecules 2017, 22, 582.
- Welch, G.N.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050.
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798.
- Perna, A.F.; Sepe, I.; Lanza, D.; Capasso, R.; Di Marino, V.; De Santo, N.G.; Ingrosso, D. The gasotransmitter hydrogen sulfide in hemodialysis patients. J. Nephrol. 2010, 23, S92–S96.
- Li, H.; Feng, S.J.; Zhang, G.Z.; Wang, S.X. Correlation of lower concentrations of hydrogen sulfide with atherosclerosis in chronic hemodialysis patients with diabetic nephropathy. Blood Purif. 2014, 38, 188–194.
- Van Guldener, C.; Stehouwer, C.D. Homocysteine metabolism in renal disease. Clin. Chem. Lab. Med. 2003, 41, 1412–1417.
- Perna, A.F.; Ingrosso, D.; Satta, E.; Lombardi, C.; Acanfora, F.; De Santo, N.G. Homocysteine metabolism in renal failure. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 53–57.
- Langan, R.C.; Goodbred, A.J. Vitamin B12 Deficiency: Recognition and Management. Am. Fam. Physician 2017, 96, 384–389.
- Brattström, L.; Wilcken, D.E. Homocysteine and cardiovascular disease: Cause or effect? Am. J. Clin. Nutr. 2000, 72, 315–323.
- Van Guldener, C.; Kulik, W.; Berger, R.; Dijkstra, D.A.; Jakobs, C.; Reijngoud, D.J.; Donker, A.J.; Stehouwer, C.D.; De Meer, K. Homocysteine and methionine metabolism in ESRD: A stable isotope study. Kidney Int. 1999, 56, 1064–1071.
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24.
- Zha, Y.; Qian, Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients 2017, 27, 208.
- Jennette, J.C.; Goldman, I.D. Inhibition of the membrane transport of folate by anions retained in uremia. J. Lab. Clin. Med. 1975, 86, 834–843.
- Bamonti-Catena, F.; Buccianti, G.; Porcella, A.; Valenti, G.; Como, G.; Finazzi, S.; Maiolo, A.T. Folate measurements in patients on regular hemodialysis treatment. Am. J. Kidney Dis. 1999, 33, 492–497.
- McMahon, G.M.; Hwang, S.J.; Tanner, R.M.; Jacques, P.F.; Selhub, J.; Muntner, P.; Fox, C.S. The association between vitamin B12, albuminuria and reduced kidney function: An observational cohort study. BMC Nephrol. 2015, 16.
- Ermens, A.A.; Vlasveld, L.T.; Lindemans, J. Significance of elevated cobalamin (vitamin B12) levels in blood. Clin. Biochem. 2003, 36, 585–590.
- Andres, E.; Serraj, K.; Zhu, J.; Vermorken, A.J. The pathophysiology of elevated vitamin B12 in clinical practice. QJM 2013, 106, 505–515.
- Koyama, K.; Yoshida, A.; Takeda, A.; Morozumi, K.; Fujinami, T.; Tanaka, N. Abnormal cyanide metabolism in uraemic patients. Nephrol. Dial. Transpl. 1997, 12, 1622–1628.
- Poddar, R.; Sivasubramanian, N.; DiBello, P.M.; Robinson, K.; Jacobsen, D.W. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: Implications for vascular disease. Circulation 2001, 103, 2717–2723.
- Zhao, J.; Chen, H.; Liu, N.; Chen, J.; Gu, Y.; Chen, J.; Yang, K. Role of Hyperhomocysteinemia and Hyperuricemia in Pathogenesis of Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2695–2699.
- Au-Yeung, K.K.; Woo, C.W.; Sung, F.L.; Yip, J.C.; Siow, Y.L.; O, K. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ. Res. 2004, 94, 28–36.
- Li, H.; Lewis, A.; Brodsky, S.; Rieger, R.; Iden, C.; Goligorsky, M.S. Homocysteine induces 3-hydroxy-3-methylglutaryl coenzyme a reductase in vascular endothelial cells: A mechanism for development of atherosclerosis? Circulation 2002, 105, 1037–1043.
- Pecoits-Filho, R.; Lindholm, B.; Stenvinkel, P. The malnutrition, inflammation, and atherosclerosis (MIA) syndrome – the heart of the matter. Nephrol. Dial. Transpl. 2002, 17, 28–31.
- Colì, L.; Donati, G.; Cappuccili, M.L.; Cianciolo, G.; Comai, G.; Cuna, V.; Carretta, E.; La Manna, G.; Stefoni, S. Role of the hemodialysis vascular access type in inflammation status and monocyte activation. Int. J. Art. Organs. 2011, 34, 481–488.
- Tsai, J.C.; Perrella, M.A.; Yoshizumi, M.; Hsieh, C.M.; Haber, E.; Schlegel, R.; Lee, M.E. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 6369–6373.
- Bottiger, A.K.; Hurtig-Wennlof, A.; Sjostrom, M.; Yngve, A.; Nilsson, T.K. Association of total plasma homocysteine with methylenetetrahydrofolate reductase genotypes 677C>T, 1298A>C, and 1793G>A and the corresponding haplotypes in Swedish children and adolescents. Int. J. Mol. Med. 2007, 19, 659–665.
- Sen, U.; Mishra, P.K.; Tyagi, N.; Tyagi, S.C. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 2010, 57, 49–58.
- Zhang, C.; Cai, Y.; Adachi, M.T.; Oshiro, S.; Aso, T.; Kaufman, R.J.; Kitajima, S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001, 276, 35867–53874.
- Baydas, G.; Reiter, R.J.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and antiapoptotic protein levels. Neuroscience 2005, 135, 879–886.
- Zhang, K.; Kaufman, R. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462.
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237.
- Perna, A.F.; Ingrosso, D.; Violetti, E.; Luciano, M.G.; Sepe, I.; Lanza, D.; Capasso, R.; Ascione, E.; Raiola, I.; Lombardi, C.; et al. Hyperhomocysteinemia in uremia—A red flag in a disrupted circuit. Semin. Dial. 2009, 22, 351–356.
- Deussen, A.; Pexa, A.; Loncar, R.; Stehr, S.N. Effects of homocysteine on vascular and tissue adenosine: A stake in homocysteine pathogenicity? Clin. Chem. Lab. Med. 2005, 43, 1007–1010.
- Solini, A.; Santini, E.; Ferrannini, E. Effect of short-term folic acid supplementation on insulin sensitivity and inflammatory markers in overweight subjects. Int. J. Obes. (Lond) 2006, 30, 1197–1202.
- Bhattacharjee, A.; Prasad, S.K.; Pal, S.; Maji, B.; Syamal, A.K.; Mukherjee, S. Synergistic protective effect of folic acid and vitamin B12 against nicotine-induced oxidative stress and apoptosis in pancreatic islets of the rat. Pharm. Biol. 2016, 54, 433–444.
- Wu, T.G.; Li, W.H.; Lin, Z.Q.; Wang, L.X. Effects of folic acid on cardiac myocyte apoptosis in rats with streptozotocin-induced diabetes mellitus. Cardiovasc. Drugs Ther. 2008, 22, 299–304.
- Verhaar, M.C.; Stroes, E.; Rabelink, T.J. Folates and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 6–13.
- Antoniades, C.; Shirodaria, C.; Warrick, N.; Cai, S.; de Bono, J.; Lee, J.; Leeson, P.; Neubauer, S.; Ratnatunga, C.; Pillai, R.; et al. 5-Methyltetrahydrofolate Rapidly Improves Endothelial Function and Decreases Superoxide Production in Human Vessels Effects on Vascular Tetrahydrobiopterin Availability and Endothelial Nitric Oxide Synthase Coupling. Circulation 2006, 114, 1193–1201.
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Payne, N.; Durrant, H.J.; Lewis, M.J.; Goodfellow, J. Folic Acid Improves Endothelial Function in Coronary Artery Disease via Mechanisms Largely Independent of Homocysteine Lowering. Circulation 2002, 105, 22–26.
- Title, L.M.; Cummings, P.M.; Giddens, K.; Genest, J.J., Jr.; Nassar, B.A. Effect of folic acid and antioxidant vitamins on endothelial dysfunction in patients with coronary artery disease. J. Am. Coll. Cardiol. 2000, 36, 758–765.
- Chambers, J.C.; Ueland, P.M.; Obeid, O.A.; Wrigley, J.; Refsum, H.; Kooner, J.S. Improved vascular endothelial function after oral B vitamins: An effect mediated through reduced concentrations of free plasma homocysteine. Circulation 2000, 102, 2479–2483.
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Lang, D.; Newcombe, R.G.; Kredan, M.B.; Lewis, M.J.; Goodfellow, J. Folate improves endothelial function in coronary artery disease: An effect mediated by reduction of intracellular superoxide? Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1196–1202.
- Pan, S.; Liu, H.; Gao, F.; Luo, H.; Lin, H.; Meng, L.; Jiang, C.; Guo, Y.; Chi, J.; Guo, H. Folic acid delays development of atherosclerosis in lowdensity lipoprotein receptor-deficient mice. J. Cell Mol. Med. 2018, 22, 3183–3191.
- Park, J.; Ahmadi, S.F.; Streja, E.; Molnar, M.Z.; Flegal, K.M.; Gillen, D.; Kovesdy, C.P.; Kalantar-Zadeh, K. Obesity paradox in end-stage kidney disease patients. Prog. Cardiovasc. Dis. 2014, 56, 415–425.
- Obi, Y.; Qader, H.; Kovesdy, C.P.; Kalantar-Zadeh, K. Latest consensus and update on proteinenergy wasting in chronic kidney disease. Curr. Opin. Clin. Nutr. Metab. Care. 2015, 18, 254–262.
- Salles, N.; Herrmann, F.; Sakbani, K.; Rapin, C.H.; Sieber, C. High vitamin B12 level: A strong predictor of mortality in elderly inpatients. J. Am. Geriatr. Soc. 2005, 53, 917–918.
- Seetharam, B.; Li, N. Transcobalamin II and its cell surface receptor. Vitam. Horm. 2000, 59, 337–366.
- Sviri, S.; Khalaila, R.; Daher, S.; Bayya, A.; Linton, D.M.; Stav, I.; van Heerden, P.V. Increased Vitamin B12 levels are associated with mortality in critically ill medical patients. Clin. Nutr. 2012, 31, 53–59.
- Bamgbola, O.F. Pattern of resistance to erythropoietin-stimulating agents in chronic kidney disease. Kidney Int. 2011, 80, 464–474.
- Saifan, C.; Samarneh, M.; Shtaynberg, N.; Nasr, R.; El-Charabaty, E.; El-Sayegh, S. Treatment of confirmed B12 deficiency in hemodialysis patients improves Epogen requirements. Int. J. Nephrol. Renovasc. Dis. 2013, 6, 89–93.
- Wu, C.C.; Zheng, C.M.; Lin, Y.F.; Lo, L.; Liao, M.T.; Lu, K.C. Role of homocysteine in end-stage renal disease. Clin. Biochem. 2012, 45, 1286–1294.
- Righetti, M.; Ferrario, G.M.; Milani, S.; Serbelloni, P.; La Rosa, L.; Uccellini, M.; Sessa, A. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med. Sci. Monit. 2003, 9, 19–24.
- Wrone, E.M.; Hornberger, J.M.; Zehnder, J.L.; McCann, L.M.; Coplon, N.S.; Fortmann, S.P. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J. Am. Soc. Nephrol. 2004, 15, 420–426.
- Zoungas, S.; McGrath, B.P.; Branley, P.; Kerr, P.G.; Muske, C.; Wolfe, R.; Atkins, R.C.; Nicholls, K.; Fraenkel, M.; Hutchison, B.G.; et al. Cardiovascular Morbidity and Mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in Chronic Renal Failure. J. Am. Coll. Cardiol. 2006, 47, 1108–1116.
- Jamison, R.L.; Hartigan, P.; Kaufman, J.S.; Goldfarb, D.S.; Warren, S.R.; Guarino, P.D.; Gaziano, J.M.; Veterans Affairs Site Investigators. Effect of Homocysteine Lowering on Mortality and Vascular Disease in Advanced Chronic Kidney Disease and End-stage Renal Disease. JAMA 2007, 298, 1163–1170.
- Heinz, J.; Kropf, S.; Domröse, U.; Westphal, S.; Borucki, K.; Luley, C.; Neumann, K.H.; Dierkes, J. B Vitamins and the Risk of Total Mortality and Cardiovascular Disease in End-Stage Renal Disease Results of a Randomized Controlled Trial. Circulation 2010, 121, 1432–1438.
- Qin, X.; Huo, Y.; Langman, C.B.; Hou, F.; Chen, Y.; Matossian, D.; Xu, X.; Wang, X. Folic acid therapy and cardiovascular disease in ESRD or advanced chronic kidney disease: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 482–488.
- Xu, X.; Qin, X.; Li, Y.; Sun, D.; Wang, J.; Liang, M.; Wang, B.; Huo, Y.; Hou, F.F.; Investigators of the Renal Substudy of the China Stroke Primary Prevention Trial (CSPPT). Efficacy of folic acid therapy on the progression of chronic kidney disease: The Renal Substudy of the China Stroke Primary Prevention Trial. JAMA Intern. Med. 2016, 176, 1443–1450.
- House, A.A.; Eliasziw, M.; Cattran, D.C.; Churchill, D.N.; Oliver, M.J.; Fine, A.; Dresser, G.K.; Spence, J.D. Effect of B-vitamin therapy on progression of diabetic nephropathy: A randomized controlled trial. JAMA 2010, 303, 1603–1609.
- Mann, J.F.; Sheridan, P.; McQueen, M.J.; Held, C.; Arnold, J.M.; Fodor, G.; Yusuf, S.; Lonn, E.M. HOPE-2 investigators. Homocysteine lowering with folic acid and B vitamins in people with chronic kidney disease—results of the renal Hope-2 study. Nephrol. Dial. Transpl. 2008, 23, 645–653.
- Vianna, A.C.; Mocelin, A.J.; Matsuo, T.; Morais-Filho, D.; Largura, A.; Delfino, V.A.; Soares, A.E.; Matni, A.M. Uremic hyperhomocysteinemia: A randomized trial of folate treatment for the prevention of cardiovascular events. Hemodial. Int. 2007, 11, 210–216.
- Righetti, M.; Serbelloni, P.; Milani, S.; Ferrario, G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif. 2006, 24, 379–386.


