Patients affected by chronic kidney disease (CKD) or end-stage renal disease (ESRD) experience a huge cardiovascular risk and cardiovascular events represent the leading causes of death. Folic acid and vitamin B12 could not only be mere cofactors in the homocysteine metabolism; they may have a direct action in determining tissue damage and cardiovascular risk.
- cardiovascular disease
- chronic kidney disease
- end-stage renal disease
- hyperhomocysteinemia
- folic acid
- vitamin B12
1. Introduction
2. B Vitamins—Homocysteine Pathway
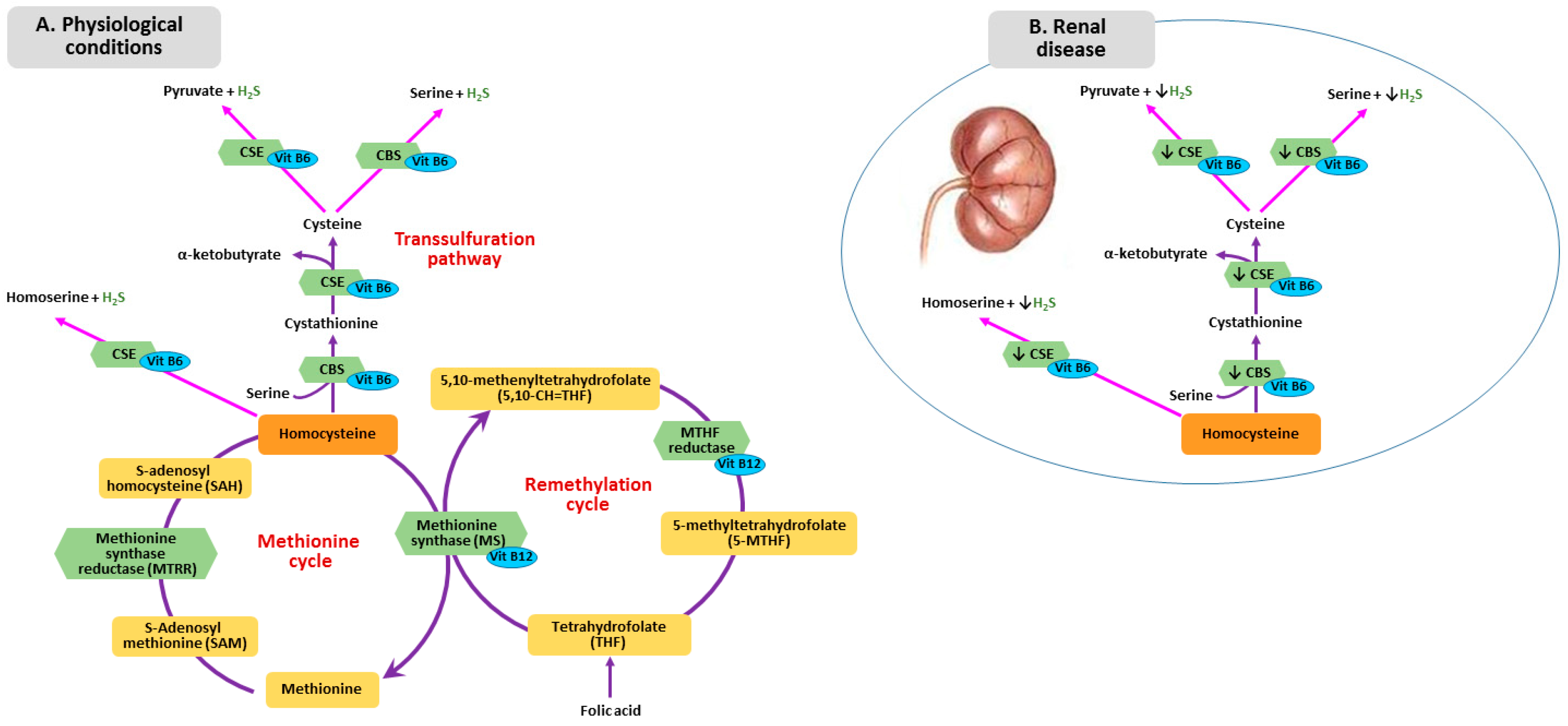
3. Metabolism of Homocysteine, Folic Acid and Vitamin B12 in CKD
4. Homocysteine-Mediated Tissue Damage
5. Folic Acid and Vitamin B12 Impairment and Tissue Injury
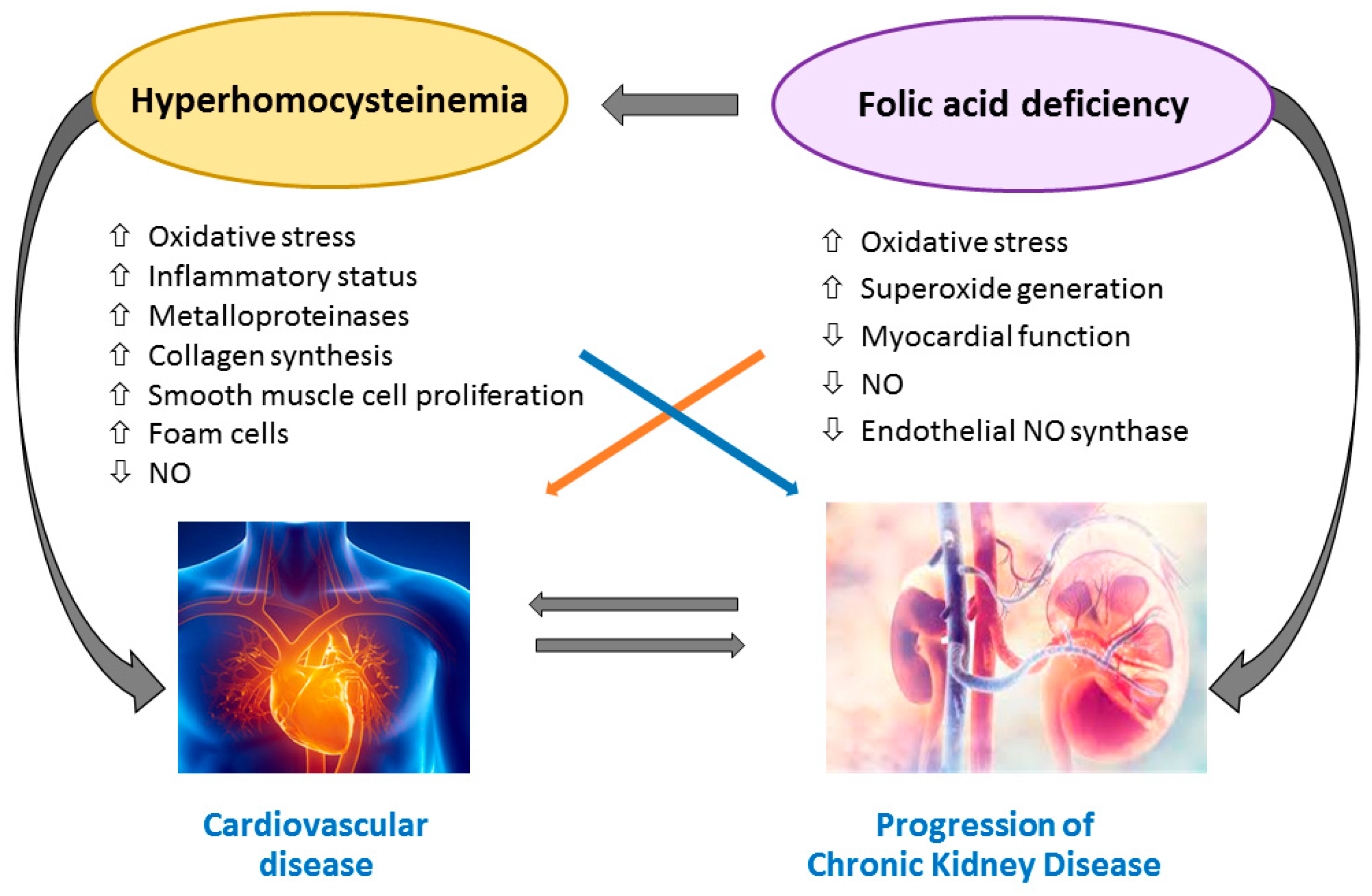
6. Effect of Folic Acid and Vitamin B12 Supplementation on CVD and Mortality in CKD and ESRD
Study | Design/Intervention | Participants, n | End Point | Follow-up, Years | Results | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
Xu et al., 2016 [87] | [79] |
Double blind RCT: enalapril 10 mg versus enalapril 10 mg plus folic acid | 15,104 (eGFR ≥ 30 mL/min). No folic acid fortification | CKD progression | 4.4 | Enalapril plus folic acid delayed CKD progression | |||||||||||
House et al., 2010 [125] | [80] |
Double blind RCT: folic acid 2.5 mg + Vitamin B6 25 mg + Vitamin B12 1 mg versus placebo | 238 (diabetic nephropathy with eGFR > 30 mL/min). Folic acid fortification | CKD progression | 2.6 | Greater GFR decrease and more CVD events in treatment group | |||||||||||
Heinz et al., 2010 [120] | [77] |
Double blind RCT: folic acid 5 mg, vitamin B12 50 µg, vitamin B6 20 mg versus placebo 3 times a week | 650 hemodialysis patients | All-cause mortality, cardiovascular events | 2 | No differences | |||||||||||
Mann et al., 2008 [126] | [81] |
Double blind RCT: folic acid 2.5 mg + vitamin B6 50 mg + vitamin B12 1 mg versus placebo | 619 CKD (eGFR <60 mL/min) | All-cause mortality, cardiovascular events | 5 | No differences | |||||||||||
Cianciolo et al., 2008 [11] | Open label randomized trial: 5-MTHF intravenous. three times a week versus folic acid 5 mg oral daily | 314 hemodialysis patients | All-cause mortality | 4.5 | Less mortality risk in 5-MTHF group (independent of homocysteine) | ||||||||||||
Jamison et al., 2007 [119] | [76] |
Double blind RCT (HOST): folic acid 40 mg + vitamin B6 100 mg + vitamin B12 2 mg versus placebo | 2056 CKD (eGFR ≤ 30) or hemodialysis (folic acid fortification) | All-cause mortality, CKD progression | 3.2 | No differences | |||||||||||
Vianna et al., 2007 [127] | [82] |
Double blind RCT: folic acid 5 mg versus placebo | 97 hemodialysis patients | Cardiovascular events | 2 | No differences | |||||||||||
Zoungas et al., 2006 [118] | [75] |
Double blind RCT (ASFAST): folic acid 15 mg versus placebo | 315 CKD (eGFR < 25 mL/min), hemodialysis and peritoneal dialysis | Cardiovascular events and mortality | 3.6 | No differences | |||||||||||
Righetti et al., 2006 [128] | [83] |
Open prospective trial: folic acid 5 mg versus untreated | 114 hemodialysis patients | Cardiovascular events | 2.4 | Folic acid decreases CVD events | |||||||||||
Wrone et al., 2004 [76] | [74] |
Three arms, double blind RCT: folic acid 1 mg or 5 mg or 15 mg | 510 hemodialysis patients | Cardiovascular events and mortality | 2 | No differences | |||||||||||
Righetti et al., 2003 [117] | [73] |
Placebo-controlled, non-blinded RCT: folic acid 5, 15, 25 mg or placebo | 81 hemodialysis patients | Cardiovascular mortality | 1 | No differences |
Abbreviations: CKD, Chronic Kidney Disease; CVD, Cardiovascular Disease; eGFR, estimated Glomerular Filtration Rate; RCT, Randomized Clinical Trial.
References
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305.
- Foley, R.N.; Parfrey, P.S.; Sarnak, M.J. Epidemiology of cardiovascular disease in chronic renal disease. J. Am. Soc. Nephrol. 1998, 9, S16–S23.
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272.
- McCullough, P.A.; Steigerwalt, S.; Tolia, K.; Chen, S.C.; Li, S.; Norris, K.C.; Whaley-Connell, A.; KEEP Investigators. Cardiovascular disease in chronic kidney disease: Data from the Kidney Early Evaluation Program (KEEP). Curr. Diab. Rep. 2011, 11, 47–55.
- Ekdahl, K.; Soveri, I.; Hilborn, J.; Fellström, B.; Nilsson, B. Cardiovascular disease in hemodialysis: Role of the intravascular innate immune system. Nat. Rev. Nephrol. 2017, 13, 285–296.
- McCully, K.S. Homocysteine and vascular disease. Nat. Med. 1996, 2, 386–389.
- Chrysant, S.G.; Chrysant, G.S. The current status of homocysteine as a risk factor for cardiovascular disease: A mini review. Expert. Rev. Cardiovasc. Ther. 2018, 16, 559–565.
- Robinson, K. Renal disease, homocysteine, and cardiovascular complications. Circulation 2004, 109, 294–295.
- Suliman, M.E.; Lindholm, B.; Barany, P.; Qureshi, A.R.; Stenvinkel, P. Homocysteine-lowering is not a primary target for cardiovascular disease prevention in chronic kidney disease patients. Semin. Dial. 2007, 20, 523–529.
- Heinz, J.; Kropf, S.; Luley, C.; Dierkes, J. Homocysteine as a risk factor for cardiovascular disease in patients treated by dialysis: A meta-analysis. Am. J. Kidney Dis. 2009, 54, 478–489.
- Cianciolo, G.; La Manna, G.; Colì, L.; Donati, G.; D’Addio, F.; Persici, E.; Comai, G.; Wratten, M.; Dormi, A.; Mantovani, V.; et al. 5-methyltetrahydrofolate administration is associated with prolonged survival and reduced inflammation in ESRD patients. Am. J. Nephrol. 2008, 28, 941–948.
- Cianciolo, G.; De Pascalis, A.; Di Lullo, L.; Ronco, C.; Zannini, C.; La Manna, G. Folic Acid and Homocysteine in Chronic Kidney Disease and Cardiovascular Disease Progression: Which Comes First? Cardiorenal. Med. 2017, 7, 255–266.
- Marti, F.; Vollenweider, P.; Marques-Vidal, P.M.; Mooser, V.; Waeber, G.; Paccaud, F.; Bochud, M. Hyperhomocysteinemia is independently associated with albuminuria in the population-based CoLaus study. BMC Public Health. 2011, 11.
- Ponte, B.; Pruijm, M.; Marques-Vidal, P.; Martin, P.Y.; Burnier, M.; Paccaud, F.; Waeber, G.; Vollenweider, P.; Bochud, M. Determinants and burden of chronic kidney disease in the population-based CoLaus study: A cross-sectional analysis. Nephrol. Dial. Transpl. 2013, 28, 2329–2339.
- Soohoo, M.; Ahmadi, S.F.; Qader, H.; Streja, E.; Obi, Y.; Moradi, H.; Rhee, C.M.; Kim, T.H.; Kovesdy, C.P.; Kalantar-Zadeh, K. Association of serum vitamin B12 and folate with mortality in incident hemodialysis patients. Nephrol. Dial. Transpl. 2017, 32, 1024–1032.
- Kang, S.S.; Wong, P.W.K.; Malinow, M.R. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu. Rev. Nutr. 1992, 12, 279–298.
- Randaccio, L.; Geremia, S.; Demitri, N.; Wuerges, J. Vitamin B12: Unique metalorganic compounds and the most complex vitamins. Molecules 2010, 15, 3228–3259.
- Long, Y.; Nie, J. Homocysteine in Renal Injury. Kidney Dis. 2016, 2, 80–87.
- Van Guldener, C.; Stam, F.; Stehouwer, C.D. Hyperhomocysteinaemia in chronic kidney disease: Focus on transmethylation. Clin. Chem. Lab. Med. 2005, 43, 1026–1031.
- Jacobsen, D.W. Homocysteine and vitamins in cardiovascular disease. Clin. Chem. 1998, 44, 1833–1843.
- Cianciolo, G.; Cappuccilli, M.; La Manna, G. The Hydrogen Sulfide-Vitamin B12-Folic Acid Axis: An Intriguing Issue in Chronic Kidney Disease. A Comment on Toohey JI: “Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer”. Molecules 2017, 22, 1216.
- Toohey, J.I. Possible Involvement of Hydrosulfide in B12-Dependent Methyl Group Transfer. Molecules 2017, 22, 582.
- Welch, G.N.; Loscalzo, J. Homocysteine and atherothrombosis. N. Engl. J. Med. 1998, 338, 1042–1050.
- Wang, R. Two’s company, three’s a crowd: Can H2S be the third endogenous gaseous transmitter? FASEB J. 2002, 16, 1792–1798.
- Perna, A.F.; Sepe, I.; Lanza, D.; Capasso, R.; Di Marino, V.; De Santo, N.G.; Ingrosso, D. The gasotransmitter hydrogen sulfide in hemodialysis patients. J. Nephrol. 2010, 23, S92–S96.
- Li, H.; Feng, S.J.; Zhang, G.Z.; Wang, S.X. Correlation of lower concentrations of hydrogen sulfide with atherosclerosis in chronic hemodialysis patients with diabetic nephropathy. Blood Purif. 2014, 38, 188–194.
- Van Guldener, C.; Stehouwer, C.D. Homocysteine metabolism in renal disease. Clin. Chem. Lab. Med. 2003, 41, 1412–1417.
- Perna, A.F.; Ingrosso, D.; Satta, E.; Lombardi, C.; Acanfora, F.; De Santo, N.G. Homocysteine metabolism in renal failure. Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 53–57.
- Langan, R.C.; Goodbred, A.J. Vitamin B12 Deficiency: Recognition and Management. Am. Fam. Physician 2017, 96, 384–389.
- Brattström, L.; Wilcken, D.E. Homocysteine and cardiovascular disease: Cause or effect? Am. J. Clin. Nutr. 2000, 72, 315–323.
- Van Guldener, C.; Kulik, W.; Berger, R.; Dijkstra, D.A.; Jakobs, C.; Reijngoud, D.J.; Donker, A.J.; Stehouwer, C.D.; De Meer, K. Homocysteine and methionine metabolism in ESRD: A stable isotope study. Kidney Int. 1999, 56, 1064–1071.
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut microbiota functions: Metabolism of nutrients and other food components. Eur. J. Nutr. 2018, 57, 1–24.
- Zha, Y.; Qian, Q. Protein Nutrition and Malnutrition in CKD and ESRD. Nutrients 2017, 27, 208.
- Jennette, J.C.; Goldman, I.D. Inhibition of the membrane transport of folate by anions retained in uremia. J. Lab. Clin. Med. 1975, 86, 834–843.
- Bamonti-Catena, F.; Buccianti, G.; Porcella, A.; Valenti, G.; Como, G.; Finazzi, S.; Maiolo, A.T. Folate measurements in patients on regular hemodialysis treatment. Am. J. Kidney Dis. 1999, 33, 492–497.
- McMahon, G.M.; Hwang, S.J.; Tanner, R.M.; Jacques, P.F.; Selhub, J.; Muntner, P.; Fox, C.S. The association between vitamin B12, albuminuria and reduced kidney function: An observational cohort study. BMC Nephrol. 2015, 16.
- Ermens, A.A.; Vlasveld, L.T.; Lindemans, J. Significance of elevated cobalamin (vitamin B12) levels in blood. Clin. Biochem. 2003, 36, 585–590.
- Andres, E.; Serraj, K.; Zhu, J.; Vermorken, A.J. The pathophysiology of elevated vitamin B12 in clinical practice. QJM 2013, 106, 505–515.
- Koyama, K.; Yoshida, A.; Takeda, A.; Morozumi, K.; Fujinami, T.; Tanaka, N. Abnormal cyanide metabolism in uraemic patients. Nephrol. Dial. Transpl. 1997, 12, 1622–1628.
- Poddar, R.; Sivasubramanian, N.; DiBello, P.M.; Robinson, K.; Jacobsen, D.W. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: Implications for vascular disease. Circulation 2001, 103, 2717–2723.
- Zhao, J.; Chen, H.; Liu, N.; Chen, J.; Gu, Y.; Chen, J.; Yang, K. Role of Hyperhomocysteinemia and Hyperuricemia in Pathogenesis of Atherosclerosis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2695–2699.
- Au-Yeung, K.K.; Woo, C.W.; Sung, F.L.; Yip, J.C.; Siow, Y.L.; O, K. Hyperhomocysteinemia activates nuclear factor-kappaB in endothelial cells via oxidative stress. Circ. Res. 2004, 94, 28–36.
- Li, H.; Lewis, A.; Brodsky, S.; Rieger, R.; Iden, C.; Goligorsky, M.S. Homocysteine induces 3-hydroxy-3-methylglutaryl coenzyme a reductase in vascular endothelial cells: A mechanism for development of atherosclerosis? Circulation 2002, 105, 1037–1043.
- Pecoits-Filho, R.; Lindholm, B.; Stenvinkel, P. The malnutrition, inflammation, and atherosclerosis (MIA) syndrome – the heart of the matter. Nephrol. Dial. Transpl. 2002, 17, 28–31.
- Colì, L.; Donati, G.; Cappuccili, M.L.; Cianciolo, G.; Comai, G.; Cuna, V.; Carretta, E.; La Manna, G.; Stefoni, S. Role of the hemodialysis vascular access type in inflammation status and monocyte activation. Int. J. Art. Organs. 2011, 34, 481–488.
- Tsai, J.C.; Perrella, M.A.; Yoshizumi, M.; Hsieh, C.M.; Haber, E.; Schlegel, R.; Lee, M.E. Promotion of vascular smooth muscle cell growth by homocysteine: A link to atherosclerosis. Proc. Natl. Acad. Sci. USA 1994, 91, 6369–6373.
- Bottiger, A.K.; Hurtig-Wennlof, A.; Sjostrom, M.; Yngve, A.; Nilsson, T.K. Association of total plasma homocysteine with methylenetetrahydrofolate reductase genotypes 677C>T, 1298A>C, and 1793G>A and the corresponding haplotypes in Swedish children and adolescents. Int. J. Mol. Med. 2007, 19, 659–665.
- Sen, U.; Mishra, P.K.; Tyagi, N.; Tyagi, S.C. Homocysteine to hydrogen sulfide or hypertension. Cell Biochem. Biophys. 2010, 57, 49–58.
- Zhang, C.; Cai, Y.; Adachi, M.T.; Oshiro, S.; Aso, T.; Kaufman, R.J.; Kitajima, S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001, 276, 35867–53874.
- Baydas, G.; Reiter, R.J.; Akbulut, M.; Tuzcu, M.; Tamer, S. Melatonin inhibits neural apoptosis induced by homocysteine in hippocampus of rats via inhibition of cytochrome c translocation and caspase-3 activation and by regulating pro- and antiapoptotic protein levels. Neuroscience 2005, 135, 879–886.
- Zhang, K.; Kaufman, R. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462.
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237.
- Perna, A.F.; Ingrosso, D.; Violetti, E.; Luciano, M.G.; Sepe, I.; Lanza, D.; Capasso, R.; Ascione, E.; Raiola, I.; Lombardi, C.; et al. Hyperhomocysteinemia in uremia—A red flag in a disrupted circuit. Semin. Dial. 2009, 22, 351–356.
- Deussen, A.; Pexa, A.; Loncar, R.; Stehr, S.N. Effects of homocysteine on vascular and tissue adenosine: A stake in homocysteine pathogenicity? Clin. Chem. Lab. Med. 2005, 43, 1007–1010.
- Solini, A.; Santini, E.; Ferrannini, E. Effect of short-term folic acid supplementation on insulin sensitivity and inflammatory markers in overweight subjects. Int. J. Obes. (Lond) 2006, 30, 1197–1202.
- Bhattacharjee, A.; Prasad, S.K.; Pal, S.; Maji, B.; Syamal, A.K.; Mukherjee, S. Synergistic protective effect of folic acid and vitamin B12 against nicotine-induced oxidative stress and apoptosis in pancreatic islets of the rat. Pharm. Biol. 2016, 54, 433–444.
- Wu, T.G.; Li, W.H.; Lin, Z.Q.; Wang, L.X. Effects of folic acid on cardiac myocyte apoptosis in rats with streptozotocin-induced diabetes mellitus. Cardiovasc. Drugs Ther. 2008, 22, 299–304.
- Verhaar, M.C.; Stroes, E.; Rabelink, T.J. Folates and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 6–13.
- Antoniades, C.; Shirodaria, C.; Warrick, N.; Cai, S.; de Bono, J.; Lee, J.; Leeson, P.; Neubauer, S.; Ratnatunga, C.; Pillai, R.; et al. 5-Methyltetrahydrofolate Rapidly Improves Endothelial Function and Decreases Superoxide Production in Human Vessels Effects on Vascular Tetrahydrobiopterin Availability and Endothelial Nitric Oxide Synthase Coupling. Circulation 2006, 114, 1193–1201.
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Payne, N.; Durrant, H.J.; Lewis, M.J.; Goodfellow, J. Folic Acid Improves Endothelial Function in Coronary Artery Disease via Mechanisms Largely Independent of Homocysteine Lowering. Circulation 2002, 105, 22–26.
- Title, L.M.; Cummings, P.M.; Giddens, K.; Genest, J.J., Jr.; Nassar, B.A. Effect of folic acid and antioxidant vitamins on endothelial dysfunction in patients with coronary artery disease. J. Am. Coll. Cardiol. 2000, 36, 758–765.
- Chambers, J.C.; Ueland, P.M.; Obeid, O.A.; Wrigley, J.; Refsum, H.; Kooner, J.S. Improved vascular endothelial function after oral B vitamins: An effect mediated through reduced concentrations of free plasma homocysteine. Circulation 2000, 102, 2479–2483.
- Doshi, S.N.; McDowell, I.F.; Moat, S.J.; Lang, D.; Newcombe, R.G.; Kredan, M.B.; Lewis, M.J.; Goodfellow, J. Folate improves endothelial function in coronary artery disease: An effect mediated by reduction of intracellular superoxide? Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1196–1202.
- Pan, S.; Liu, H.; Gao, F.; Luo, H.; Lin, H.; Meng, L.; Jiang, C.; Guo, Y.; Chi, J.; Guo, H. Folic acid delays development of atherosclerosis in lowdensity lipoprotein receptor-deficient mice. J. Cell Mol. Med. 2018, 22, 3183–3191.
- Park, J.; Ahmadi, S.F.; Streja, E.; Molnar, M.Z.; Flegal, K.M.; Gillen, D.; Kovesdy, C.P.; Kalantar-Zadeh, K. Obesity paradox in end-stage kidney disease patients. Prog. Cardiovasc. Dis. 2014, 56, 415–425.
- Obi, Y.; Qader, H.; Kovesdy, C.P.; Kalantar-Zadeh, K. Latest consensus and update on proteinenergy wasting in chronic kidney disease. Curr. Opin. Clin. Nutr. Metab. Care. 2015, 18, 254–262.
- Salles, N.; Herrmann, F.; Sakbani, K.; Rapin, C.H.; Sieber, C. High vitamin B12 level: A strong predictor of mortality in elderly inpatients. J. Am. Geriatr. Soc. 2005, 53, 917–918.
- Seetharam, B.; Li, N. Transcobalamin II and its cell surface receptor. Vitam. Horm. 2000, 59, 337–366.
- Sviri, S.; Khalaila, R.; Daher, S.; Bayya, A.; Linton, D.M.; Stav, I.; van Heerden, P.V. Increased Vitamin B12 levels are associated with mortality in critically ill medical patients. Clin. Nutr. 2012, 31, 53–59.
- Bamgbola, O.F. Pattern of resistance to erythropoietin-stimulating agents in chronic kidney disease. Kidney Int. 2011, 80, 464–474.
- Saifan, C.; Samarneh, M.; Shtaynberg, N.; Nasr, R.; El-Charabaty, E.; El-Sayegh, S. Treatment of confirmed B12 deficiency in hemodialysis patients improves Epogen requirements. Int. J. Nephrol. Renovasc. Dis. 2013, 6, 89–93.
- Wu, C.C.; Zheng, C.M.; Lin, Y.F.; Lo, L.; Liao, M.T.; Lu, K.C. Role of homocysteine in end-stage renal disease. Clin. Biochem. 2012, 45, 1286–1294.
- Righetti, M.; Ferrario, G.M.; Milani, S.; Serbelloni, P.; La Rosa, L.; Uccellini, M.; Sessa, A. Effects of folic acid treatment on homocysteine levels and vascular disease in hemodialysis patients. Med. Sci. Monit. 2003, 9, 19–24.
- Wrone, E.M.; Hornberger, J.M.; Zehnder, J.L.; McCann, L.M.; Coplon, N.S.; Fortmann, S.P. Randomized trial of folic acid for prevention of cardiovascular events in end-stage renal disease. J. Am. Soc. Nephrol. 2004, 15, 420–426.
- Zoungas, S.; McGrath, B.P.; Branley, P.; Kerr, P.G.; Muske, C.; Wolfe, R.; Atkins, R.C.; Nicholls, K.; Fraenkel, M.; Hutchison, B.G.; et al. Cardiovascular Morbidity and Mortality in the Atherosclerosis and Folic Acid Supplementation Trial (ASFAST) in Chronic Renal Failure. J. Am. Coll. Cardiol. 2006, 47, 1108–1116.
- Jamison, R.L.; Hartigan, P.; Kaufman, J.S.; Goldfarb, D.S.; Warren, S.R.; Guarino, P.D.; Gaziano, J.M.; Veterans Affairs Site Investigators. Effect of Homocysteine Lowering on Mortality and Vascular Disease in Advanced Chronic Kidney Disease and End-stage Renal Disease. JAMA 2007, 298, 1163–1170.
- Heinz, J.; Kropf, S.; Domröse, U.; Westphal, S.; Borucki, K.; Luley, C.; Neumann, K.H.; Dierkes, J. B Vitamins and the Risk of Total Mortality and Cardiovascular Disease in End-Stage Renal Disease Results of a Randomized Controlled Trial. Circulation 2010, 121, 1432–1438.
- Qin, X.; Huo, Y.; Langman, C.B.; Hou, F.; Chen, Y.; Matossian, D.; Xu, X.; Wang, X. Folic acid therapy and cardiovascular disease in ESRD or advanced chronic kidney disease: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2011, 6, 482–488.
- Xu, X.; Qin, X.; Li, Y.; Sun, D.; Wang, J.; Liang, M.; Wang, B.; Huo, Y.; Hou, F.F.; Investigators of the Renal Substudy of the China Stroke Primary Prevention Trial (CSPPT). Efficacy of folic acid therapy on the progression of chronic kidney disease: The Renal Substudy of the China Stroke Primary Prevention Trial. JAMA Intern. Med. 2016, 176, 1443–1450.
- House, A.A.; Eliasziw, M.; Cattran, D.C.; Churchill, D.N.; Oliver, M.J.; Fine, A.; Dresser, G.K.; Spence, J.D. Effect of B-vitamin therapy on progression of diabetic nephropathy: A randomized controlled trial. JAMA 2010, 303, 1603–1609.
- Mann, J.F.; Sheridan, P.; McQueen, M.J.; Held, C.; Arnold, J.M.; Fodor, G.; Yusuf, S.; Lonn, E.M. HOPE-2 investigators. Homocysteine lowering with folic acid and B vitamins in people with chronic kidney disease—results of the renal Hope-2 study. Nephrol. Dial. Transpl. 2008, 23, 645–653.
- Vianna, A.C.; Mocelin, A.J.; Matsuo, T.; Morais-Filho, D.; Largura, A.; Delfino, V.A.; Soares, A.E.; Matni, A.M. Uremic hyperhomocysteinemia: A randomized trial of folate treatment for the prevention of cardiovascular events. Hemodial. Int. 2007, 11, 210–216.
- Righetti, M.; Serbelloni, P.; Milani, S.; Ferrario, G. Homocysteine-lowering vitamin B treatment decreases cardiovascular events in hemodialysis patients. Blood Purif. 2006, 24, 379–386.