 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | ANTONELLA CACCHIONE | + 1553 word(s) | 1553 | 2021-09-22 11:43:30 | | | |
| 2 | Amina Yu | + 34 word(s) | 1587 | 2021-10-08 05:16:51 | | |
Video Upload Options
Pediatric spinal tumors are rare and account for 10% of all central nervous system tumors in children. Onset usually occurs with chronic nonspecific symptoms and may depend on the intra- or extradural neoplastic location. Meningiomas, schwannomas, and neurofibromas are the most common intradural-extramedullary lesions, while astrocytomas and ependymomas represent the majority of intramedullary tumors.
1. Introduction
Pediatric spinal tumors are relatively rare and account for 10% of all central nervous system tumors in children [1][2][3]. Related to location, spinal neoplasms are divided into extradural (two-thirds of cases), intradural-extramedullary, and intramedullary (one-third of cases) lesions [3]. Extradural neoplasms have a variable origin and arise from bone, soft tissues, and meningeal sheets; intradural-extramedullary neoplasms are usually meningiomas, schwannomas, and neurofibromas. Intramedullary cancers mainly include glial tumors such as astrocytomas and ependymomas, which represent 90% of all intramedullary neoplasms [4], but can also include gangliogliomas and hemangioblastomas.
Computed tomography (CT) and radiographic imaging can be useful to evaluate bone involvement and spinal stability in selected cases.
The advancement of molecular findings in pediatric central nervous system tumors (CNS) provides additional information regarding the tumor subtypes and their biological behaviors and patient outcomes. These novel findings help to reach a detailed diagnosis, obtaining new therapeutic options by targeted drugs. These molecular advances have also been described for spinal neoplasms.
Our aim in this review was to describe the imaging findings of the most frequent intradural intra/extramedullary pediatric spinal tumors according to the World Health Organization (WHO) 2016 revised classification [5] and to investigate the connection with the latest molecular findings and genetic syndromes. It is also interesting to mention some very rare spinal tumors that can be exceptionally diagnosed in children, such as the diffuse leptomeningeal glioneuronal tumor (DL-GNT), due to its typical radiological aspect.
2. Gliomas and Mixed Neuronal–Glial Tumors
The genetic landscape of pediatric spinal tumors has been less studied than that of intracranial localization: the involvement of KIAA1549–BRAF (v-raf murine sarcoma viral oncogene homolog B1), BRAFV600E, PTPN11, H3F3A, TP53, FGFR1, and CDKN2A deletion has been detected in pediatric spinal tumors [6][7]. One of the most studied molecular alterations concerns LGG and it is KIAA1549-BRAF fusion that causes the hyperactivation of the MAPK/ERK pathway.
It has been largely investigated and demonstrated that KIAA1549-BRAF-fused cerebellar pLGGs have a better prognosis compared to the ones not carrying the fusion; the same evidence has been described for the spinal low-grade gliomas [6][8]. On the other hand, the presence of BRAF fusion in the tumor suggests the potential use of target therapy, such as MEK inhibitors [9], while the use of BRAF inhibitors are indicated and useful when the BRAFV600 mutation is present [10]. However, the association between KIAA1549-BRAF fusion and outcome has not yet been validated, as according to some studies, this molecular alteration could not predict the prognosis [11]. Mutations in genes encoding histone H3.1 (HIST1H3B) and H3.3 (H3F3A) have been considered as a hallmark of diffuse midline gliomas. Instead, histonic mutations are also rarely found in pediatric low-grade gliomas [12]; this concept is important in highlighting the pathological heterogeneity in gliomas and the overlapped genetic landscape between high- and low-grade gliomas [6][13].
In the landscape of spinal pediatric LGG, clinical outcome can be related to type of resection, age, and presence of metastasis: there is a known link between subtotal resection, young age, and metastatic spreading at diagnosis with less event-free survival (EFS). A primary management goal for pediatric spinal tumors is to extend long-term follow-up because progression is possible even 10–20 years after diagnosis; for this reason, it is important to expand our knowledge around these tumors [11].
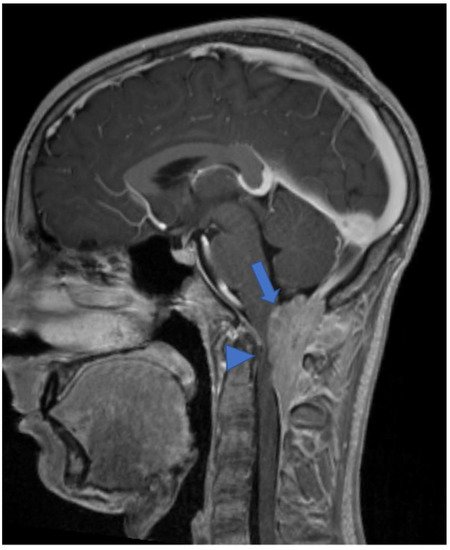
DL-GNT is a rare neuronal–glial tumor that was first defined in 2016. It is more common in males, with a median age at diagnosis of 4–6 years [5], characterized by slow growth and leptomeningeal dissemination. MRI may show different features, such as multiple spinal and/or intracranial (i) leptomeningeal nodular lesions with diffuse sheet enhancement (the most frequent findings), (ii) cystic-like leptomeningeal nodular lesions (T2 hyperintensity), (iii) intraparenchymal lesions [14][15], (iv) an intraparenchymal spinal/intracranial mass, and (v) nonenhancing cystic-like small intra-axial lesions ( Figure 2 ) [14]. Differential diagnoses include infections, leptomeningeal dissemination of other neoplasms, lymphoproliferative diseases, and neurosarcoidosis (rare in pediatric age).
3. Meningiomas and Tumors of the Paraspinal Nerves
Pediatric schwannomas develop from Schwann cells and are rarely sporadic. They account for 0.3% of intraspinal tumors [16] and are most commonly associated with NF2. When their localization is the spine, they may arise from the intra- (more common) or extradural tract of the spinal root with variable associated signs and symptoms, such as bone remodeling. The most important feature is that the lesion usually shows expansive (and non-infiltrative) growth relative to the nerve root, a well-defined capsule, and a plane of cleavage [3]. In the T1- and T2-weighted sequences, the tumor appears iso-/hypo-intense and iso-/hyper-intense, respectively, with homogeneous enhancement [3]. Differential diagnoses of spinal schwannomas are those of plexiform neurofibromas, which are intradural-extramedullary tumors. With similar signaling and enhancement features, neurofibromas differ from schwannomas in that they include both Schwann cells and fibroblasts, have infiltrative rather than expansive growth towards the spinal root [3], and could have a malignant evolution with rapid development [17]. Sometimes in neurofibromas, it is possible to observe the “target sign” (central area of hypointensity in T2) due to the high stromal component of collagen [17]. In pediatric patients with malignant peripheral nerve sheath tumors, the 5-year event-free survival (EFS) has been shown to be around 52.9% and overall survival (OS) is around 62.1% [18].
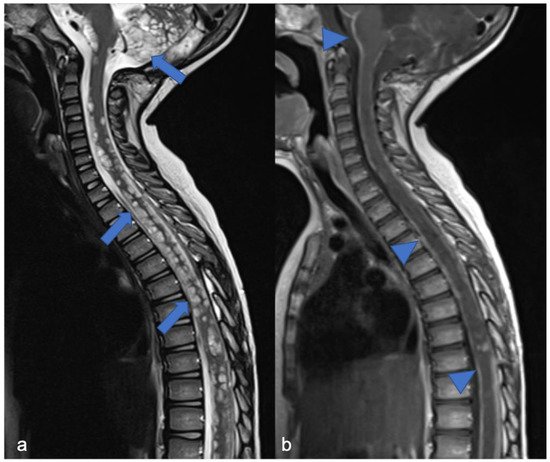
Meningiomas, rare in children, make up only 3% of pediatric SNC tumors [19] and are closely related to the diagnosis of NF2; it is estimated that 20% of NF2 patients harbor spinal meningiomas. Meningiomas in NF2 are typically WHO grade 1, slow-growing, benign tumors. When present, meningiomas in NF2 patients are often multiple, which contributes significantly to morbidity and mortality. The “clear cell meningioma (CCM)” (WHO grade II) is a typical pediatric/juvenile spinal meningioma and represents the most common histological subtype of sporadic pediatric spinal meningioma [3]. They are characterized by early local recurrence and cerebrospinal fluid metastasis. The World Health Organization defines CCM as a grade II cancer. Its incidence rate in children is higher than that in adults, who may manifest aggressive features such as recurrence and CSF dissemination [20][21][22][23]. The MRI features include dural tail, isointensity in T1 and T2 compared to the spinal cord, clear margins, and homogeneous enhancement ( Figure 2 ) [3]. Outcome is closely related to surgery: gross total resection is associated with a good clinical outcome, while subtotal resection or partial resection could be associated with recurrence or growth of the residual lesion with clinical progression even 60 months after the first surgery (long-term recurrence) [24].
Some authors suggest a complete clinical, radiological, and genetic evaluation in single, apparently sporadic, cranial/spinal meningioma and schwannoma [16]. Beyond the NF2, the SMARCE1 (SWI/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily E, member 1) gene mutation was found in young adults (16–24 years) with meningioma. Heterozygous loss-of-function mutations in the SWI/SNF chromatin-remodeling complex subunit gene SMARCE1 play a key role in the pathogenesis of spinal meningiomas with clear cell histology [25]. Furthermore, in young people (1–15 years), the germline LZTR1-mutation is associated with the development of schwannomas, as well as that of SMARCB1, which, in the older age group, accounts for 25% of schwannomatosis disease [16].
4. Embryonal Tumors
Embryonal tumors with spinal localization are very rare. In our opinion, among these, it is useful to describe the atypical teratoid/rhabdoid tumors and embryonal tumors with multilayered rosettes, which are rarely found in children.
Atypical teratoid/rhabdoid tumor (ATRT) is a rare brain tumor that is more common in children under 2 years of age and accounts for 1–2% of all pediatric brain neoplasms [25][26][27] with aggressive development and poor prognosis [27][28]. Intramedullary spinal atypical teratoid/rhabdoid tumor (spATRT) origin accounts for 3.5% of all ATRTs, and intradural-extramedullary and extradural cases have also been described. MRIs show an inhomogeneous lesion and iso/hypointense in T1 and T2 due to the presence of solid and/or cystic components, necrosis, and bleeding with heterogeneous enhancement [3][26]. There are three molecular subgroups based on DNA methylation, which include ATRT-MYC, ATRT-SHH, and ATRT-TYR [29][30], but currently there is no recognized correlation with localization, response to treatment, and prognosis due to the few cases of spATRT [31]. Moreover, the deletion or mutation of the SMARCB1 locus, a typical genetic alteration, was detected [26][32][33]. Outcome of children with ATRT is poor and the median survival is about 17 months [34][35].
Embryonal tumor with multilayered rosettes (ETMR), C19MC-altered, is a rare and aggressive, high-grade neoplasm, more common in infants (<3 years old) [36][37], with an average survival of about 1 year [38]. Introduced in 2016, ETMR encompasses previous diagnostic entities, such as embryonal tumor with abundant neuropil and true rosettes (ETANTR) [39], ependymoblastoma (EBL), and medulloepithelioma (MEPL) [5][40]. ETMRs have frequent brain localization (70% supratentorial; 30% infratentorial) and are exceptionally pronounced in the spinal cord (<1%) [41]. MRIs show a large tumor with restricted diffusion, cystic components, and heterogeneous pre-contrast signal and enhancement [42][43]. The genetic hallmark of this neoplasm is the C19MC amplification (90% of all ETMRs), and it is possible to identify (in the rosette-forming cells) LIN28A-positive immunostaining (RNA-binding protein) [43]. Half of the ETMR cases that do not show C19MC amplification are characterized by biallelic DICER1 inactivation [41].
References
- Koeller, K.K.; Rosenblum, R.S.; Morrison, A.L. Neoplasms of the spinal cord and filum terminale: Radiologic-Pathologic correlation. RadioGraphics 2000, 20, 1721–1749.
- Wilson, P.E.; Oleszek, J.L.; Clayton, G.H. Pediatric spinal cord tumors and masses. J. Spinal Cord Med. 2007, 30, S15–S20.
- Rossi, A.; Gandolfo, C.; Morana, G.; Tortori-Donati, P. Tumors of the Spine in children. Neuroimaging Clin. N. Am. 2007, 17, 17–35.
- Huisman, T.A.G.M. Pediatric tumors of the spine. Cancer Imaging 2009, 9, S45–S48.
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A Summary. Acta Neuropathol. 2016, 131, 803–820.
- Grob, S.T.; Nobre, L.; Campbell, K.R.; Davies, K.D.; Ryall, S.; Aisner, D.L.; Hoffman, L.; Zahedi, S.; Morin, A.; Crespo, M.; et al. Clinical and Molecular characterization of a multi-institutional cohort of pediatric spinal cord low-grade gliomas. Neurooncol. Adv. 2020, 2, vdaa103.
- Konovalov, N.A.; Asyutin, D.S.; Shayhaev, E.G.; Kaprovoy, S.V.; Timonin, S.Y. Molecular Biomarkers of brain and spinal cord astrocytomas. Acta Nat. 2019, 11, 17–27.
- Hawkins, C.; Walker, E.; Mohamed, N.; Zhang, C.; Jacob, K.; Shirinian, M.; Alon, N.; Kahn, D.; Fried, I.; Scheinemann, K.; et al. BRAF-KIAA1549 Fusion Predicts Better Clinical Outcome in Pediatric Low-Grade Astrocytoma. Clin. Cancer Res. 2011, 17, 4790–4798.
- Banerjee, A.; Jakacki, R.I.; Onar-Thomas, A.; Wu, S.; Nicolaides, T.; Young Poussaint, T.; Fangusaro, J.; Phillips, J.; Perry, A.; Turner, D.; et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric Patients with recurrent or refractory low-grade glioma: A Pediatric brain tumor consortium (PBTC) study. Neuro-Oncol. 2017, 19, 1135–1144.
- Schindler, G.; Capper, D.; Meyer, J.; Janzarik, W.; Omran, H.; Herold-Mende, C.; Schmieder, K.; Wesseling, P.; Mawrin, C.; Hasselblatt, M.; et al. Analysis of BRAF V600E mutation in 1320 Nervous system tumors reveals high mutation frequencies in pleomorphic xanthoastrocytoma, ganglioglioma and extra-cerebellar pilocytic astrocytoma. Acta Neuropathol. 2011, 121, 397–405.
- Perwein, T.; Benesch, M.; Kandels, D.; Pietsch, T.; Schmidt, R.; Quehenberger, F.; Bison, B.; Warmuth-Metz, M.; Timmermann, B.; Krauss, J.; et al. High frequency of disease progression in pediatric spinal cord low-grade glioma (lgG): Management Strategies and results from the German LGG Study Group. Neuro-Oncol. 2020, 23, 1148–1162.
- Okuda, T.; Hata, N.; Suzuki, S.O.; Yoshimoto, K.; Arimura, K.; Amemiya, T.; Akagi, Y.; Kuga, D.; Oba, U.; Koga, Y.; et al. Pediatric ganglioglioma with an H3 K27M mutation arising from the cervical spinal cord. Neuropathology 2018, 38, 422–427.
- Chai, R.-C.; Zhang, Y.-W.; Liu, Y.-Q.; Chang, Y.-Z.; Pang, B.; Jiang, T.; Jia, W.-Q.; Wang, Y.-Z. The molecular characteristics of spinal cord gliomas with or without H3 K27M mutation. Acta Neuropathol. Commun. 2020, 8, 40.
- Gardiman, M.P.; Fassan, M.; Orvieto, E.; D’Avella, D.; Denaro, L.; Calderone, M.; Severino, M.; Scarsello, G.; Viscardi, E.; Perilongo, G. Diffuse Leptomeningeal glioneuronal tumors: A new entity? Brain Pathol. 2010, 20, 361–366.
- Chiang, J.C.H.; Harreld, J.H.; Orr, B.A.; Sharma, S.; Ismail, A.; Segura, A.D.; Ellison, D.W. Low-Grade spinal glioneuronal tumors with BRAF Gene fusion and 1p deletion but without leptomeningeal dissemination. Acta Neuropathol. 2017, 134, 159–162.
- Pathmanaban, O.N.; Sadler, K.V.; Kamaly-Asl, I.D.; King, A.T.; Rutherford, S.A.; Hammerbeck-Ward, C.; McCabe, M.G.; Kilday, J.-P.; Beetz, C.; Poplawski, N.K.; et al. Association of genetic predisposition with solitary schwannoma or meningioma in children and young adults. JAMA Neurol. 2017, 74, 1123–1129.
- Soderlund, K.A.; Smith, A.B.; Rushing, E.J.; Smirniotopolous, J.G. Radiologic-Pathologic correlation of pediatric and adolescent spinal neoplasms: Part 2, Intradural extramedullary spinal neoplasms. Am. J. Roentgenol. 2012, 198, 44–51.
- Van Noesel, M.M.; Orbach, D.; Brennan, B.; Kelsey, A.; Zanetti, I.; de Salvo, G.L.; Gaze, M.N.; Craigie, R.J.; McHugh, K.; Francotte, N.; et al. Outcome and prognostic factors in pediatric malignant peripheral nerve sheath tumors: An analysis of the European Pediatric Soft Tissue Sarcoma Group (EpSSG) NRSTS-2005 prospective study. Pediatr. Blood Cancer 2019, 66, e27833.
- Balogun, J.A.; Halliday, W.; Bouffet, E.; Kulkarni, A.V. Spinal Clear Cell Meningioma in a 3-Year-Old: A Case Report. Pediatric Neurosurg. 2013, 49, 311–315.
- Alameda, F.; Lloreta, J.; Ferrer, M.D.; Corominas, J.M.; Galitó, E.; Serrano, S. Clear cell meningioma of the lumbo-sacral spine with chordoid features. Ultrastruct. Pathol. 1999, 23, 51–58.
- Lee, W.; Chang, K.-H.; Choe, G.; Chi, J.G.; Chung, C.-K.; Kim, I.H.; Han, M.H.; Park, S.-W.; Shin, S.J.; Koh, Y.H. MR Imaging features of clear-cell meningioma with diffuse leptomeningeal seeding. Am. J. Neuroradiol. 2000, 21, 130–132.
- Yu, K.B.; Lim, M.K.; Kim, H.J.; Suh, C.H.; Park, H.C.; Kim, E.Y.; Han, H.S. Clear-Cell meningioma: CT and MR Imaging findings in two cases involving the spinal canal and cerebellopontine angle. Korean J. Radiol. 2002, 3, 125–129.
- Clear Cell Meningioma. A clinicopathologic study of a potentially aggressive variant of meningioma. Am. J. Surg. Pathol. 1995, 19, 493–505.
- Wu, L.; Yang, C.; Liu, T.; Fang, J.; Yang, J.; Xu, Y. Clinical Features and long-term outcomes of pediatric spinal meningiomas. J. Neurooncol. 2017, 133, 347–355.
- Smith, M.J.; O’Sullivan, J.; Bhaskar, S.S.; Hadfield, K.D.; Poke, G.; Caird, J.; Sharif, S.; Eccles, D.; Fitzpatrick, D.; Rawluk, D.; et al. Loss-of-Function mutations in SMARCE1 Cause an inherited disorder of multiple spinal meningiomas. Nat. Genet. 2013, 45, 295–298.
- Biswas, A.; Kashyap, L.; Kakkar, A.; Sarkar, C.; Julka, P.K. Atypical Teratoid/rhabdoid tumors: Challenges and search for solutions. Cancer Manag. Res. 2016, 8, 115–125.
- Rickert, C.H.; Paulus, W. Epidemiology of central nervous system tumors in childhood and adolescence based on the new WHO classification. Childs Nerv. Syst. 2001, 17, 503–511.
- Woehrer, A.; Slavc, I.; Waldhoer, T.; Heinzl, H.; Zielonke, N.; Czech, T.; Benesch, M.; Hainfellner, J.A.; Haberler, C. Austrian Brain Tumor Registry incidence of atypical teratoid/rhabdoid tumors in children: A population-based study by the Austrian Brain Tumor Registry, 1996–2006. Cancer 2010, 116, 5725–5732.
- Frühwald, M.C.; Biegel, J.A.; Bourdeaut, F.; Roberts, C.W.M.; Chi, S.N. Atypical Teratoid/rhabdoid tumors—Current concepts, advances in biology, and potential future therapies. Neuro-Oncol. 2016, 18, 764–778.
- Johann, P.D.; Erkek, S.; Zapatka, M.; Kerl, K.; Buchhalter, I.; Hovestadt, V.; Jones, D.T.W.; Sturm, D.; Hermann, C.; Segura Wang, M.; et al. Atypical Teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 2016, 29, 379–393.
- Benesch, M.; Nemes, K.; Neumayer, P.; Hasselblatt, M.; Timmermann, B.; Bison, B.; Ebetsberger-Dachs, G.; Bourdeaut, F.; Dufour, C.; Biassoni, V.; et al. Spinal cord atypical teratoid/rhabdoid tumors in children: Clinical, genetic, and outcome characteristics in a Representative European Cohort. Pediatr. Blood Cancer 2020, 67, e28022.
- Biegel, J.A.; Tan, L.; Zhang, F.; Wainwright, L.; Russo, P.; Rorke, L.B. Alterations of the HSNF5/INI1 Gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin. Cancer Res. 2002, 8, 3461–3467.
- Chamberlain, M.C.; Tredway, T.L. Adult primary intradural spinal cord tumors: A review. Curr. Neurol. Neurosci. Rep. 2011, 11, 320–328.
- Ginn, K.F.; Gajjar, A. Atypical Teratoid rhabdoid tumor: Current therapy and future directions. Front. Oncol. 2012, 2, 114.
- Das, J.M.; Abraham, M.; Nandeesh, B.N.; Nair, S.N. Pediatric Suprasellar atypical teratoid rhabdoid tumor arising from the third ventricle: A Rare tumor at a very rare location. Asian J. Neurosurg. 2018, 13, 873–876.
- Korshunov, A.; Sturm, D.; Ryzhova, M.; Hovestadt, V.; Gessi, M.; Jones, D.T.W.; Remke, M.; Northcott, P.; Perry, A.; Picard, D.; et al. Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol. 2014, 128, 279–289.
- Spence, T.; Sin-Chan, P.; Picard, D.; Barszczyk, M.; Hoss, K.; Lu, M.; Kim, S.-K.; Ra, Y.-S.; Nakamura, H.; Fangusaro, J.; et al. CNS-PNETs with C19MC amplification and/or LIN28 Expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol. 2014, 128, 291–303.
- Mayr, L.; Gojo, J.; Peyrl, A.; Azizi, A.A.; Stepien, N.M.; Pletschko, T.; Czech, T.; Dorfer, C.; Lambo, S.; Dieckmann, K.; et al. Potential importance of early focal radiotherapy following gross total resection for long-term survival in children with embryonal tumors with multilayered rosettes. Front. Oncol. 2020, 10, 584681.
- Eberhart, C.G.; Brat, D.J.; Cohen, K.J.; Burger, P.C. Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr. Dev. Pathol. 2000, 3, 346–352.
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO Classification of Tumours of the Central Nervous System. Acta Neuropathol. 2007, 114, 97–109.
- Lambo, S.; von Hoff, K.; Korshunov, A.; Pfister, S.M.; Kool, M. ETMR: A Tumor entity in its infancy. Acta Neuropathol. 2020, 140, 249–266.
- Nowak, J.; Seidel, C.; Berg, F.; Pietsch, T.; Friedrich, C.; von Hoff, K.; Rutkowski, S.; Warmuth-Metz, M. MRI characteristics of ependymoblastoma: Results from 22 centrally reviewed cases. Am. J. Neuroradiol. 2014, 35, 1996–2001.
- Dangouloff-Ros, V.; Tauziède-Espariat, A.; Roux, C.-J.; Levy, R.; Grévent, D.; Brunelle, F.; Gareton, A.; Puget, S.; Beccaria, K.; Blauwblomme, T.; et al. CT and multimodal MR Imaging features of embryonal tumors with multilayered rosettes in children. Am. J. Neuroradiol. 2019, 40, 32–736.

