Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ming-Tso Yan | + 3098 word(s) | 3098 | 2021-09-26 10:32:15 | | | |
| 2 | Conner Chen | Meta information modification | 3098 | 2021-10-08 06:08:22 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Yan, M. Retard Progression of CKD. Encyclopedia. Available online: https://encyclopedia.pub/entry/14767 (accessed on 04 August 2026).
Yan M. Retard Progression of CKD. Encyclopedia. Available at: https://encyclopedia.pub/entry/14767. Accessed August 04, 2026.
Yan, Ming-Tso. "Retard Progression of CKD" Encyclopedia, https://encyclopedia.pub/entry/14767 (accessed August 04, 2026).
Yan, M. (2021, September 30). Retard Progression of CKD. In Encyclopedia. https://encyclopedia.pub/entry/14767
Yan, Ming-Tso. "Retard Progression of CKD." Encyclopedia. Web. 30 September, 2021.
Copy Citation
Chronic kidney disease (CKD), defined as the presence of irreversible structural or functional kidney damages, increases the risk of poor outcomes due to its association with multiple complications, including altered mineral metabolism, anemia, metabolic acidosis, and increased cardiovascular events. The mainstay of treatments for CKD lies in the prevention of the development and progression of CKD as well as its complications.
acute kidney injury
chronic kidney disease
renal progression
therapy for renal failure
1. Introduction
Chronic kidney disease (CKD) is defined as a progressive and irreversible loss of renal function evidenced by an estimated glomerular filtration rate (eGFR) of <60 mL/min per 1.73m2, the persistent presence of manifestations that are suggestive of kidney damage (proteinuria, active urine sediments, histological damages, structural abnormalities or a history of kidney transplantation), or both, lasting for more than 3 months [1]. CKD has long been a worldwide public health concern and constitutes a heavy healthcare and economic burden, as a reduced GFR is widely known to increase the risk of cardiovascular events, hospitalization, cognitive dysfunction, and overall mortality [2]. The prevalence of CKD varies according to geographic areas, mostly ranging from 10 to 20%, but rises gradually, particularly in developed countries [3][4][5]. This trend can be partially attributed to the expanding aging population globally [6]. In addition, the increased prevalence of risk factors such as diabetes mellitus (DM), hypertension, and obesity in patients with CKD is also notable [7][8].
Having a diagnosis of CKD means that an individual’s renal function has entered into a “point of no return,” indicating that the deterioration of renal function over time is inevitable and frequently irreversible. However, there can still be different patterns of renal function decline in patients with CKD. These patterns can be classified intuitively into very fast, fast, moderate, or slow, depending on the threshold required for defining the renal function decline rate, using mL/min/year or mL/min/month (Figure 1A). These differences in patterns largely reflect the heterogeneity of CKD origins and the subsequent pathologies, adjunct comorbidities, interventions that patients receive, and other harsh environmental exposures [9]. From this perspective, finding how to retard renal progression in patients with CKD is still fraught with challenges.
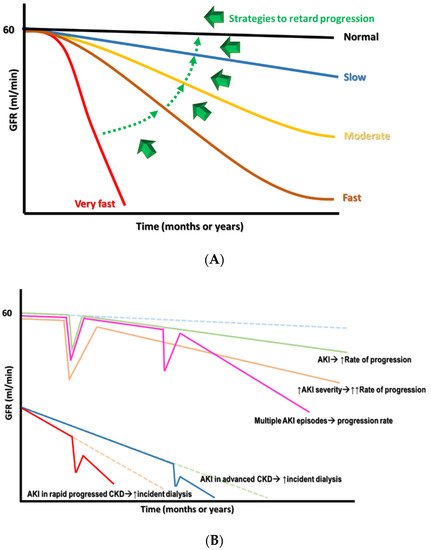
Figure 1. (A) The different declined rates of renal function in CKD with the target switch from superfast to slow rate. (B) The consequence of AKI on CKD progression, depending on the severity and frequency of episodes.
2. Risks of CKD Progression
Estimated GFR trajectories are highly variable in CKD. This phenomenon implies that a wide array of heterogeneous risk factors may contribute to CKD progression and that these risk factors may be potential therapeutic targets if we wish to achieve renoprotection. Socioeconomic factors and lifestyle factors (e.g., diet, sleep deprivation, smoking, and lack of exercise) are well-known risk factors that are associated with CKD progression. Systemic and metabolic disorders, including DM, hypertension, gout, and cardiovascular diseases, can also precipitate the development of CKD and aggravate eGFR decline (Figure 2) [10]. Recently, atrial fibrillation (AF) has also been shown to be a contributor to rapid eGFR decline, and the CHA2DS2-VASc score, a stroke-risk stratification model for patients with AF, can predict renal progression [11].
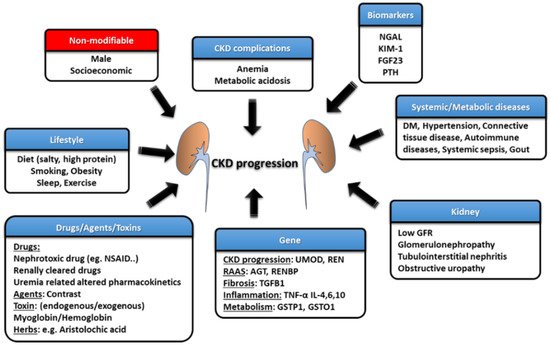
Figure 2. The risk factors and management strategies of CKD development and progression.
The intrinsic factors that are related to kidneys per se also play an important role in influencing renal function decline, such as GFR, proteinuria, glomerulopathy, interstitial lesions, and renal outlet obstruction (obstructive nephropathy). Each glomerulopathy distinctly influences the pace of renal function deterioration; for example, focal segmental glomerulosclerosis (FSGS) is more likely to result in a faster GFR decline, while the rate is lower for patients with IgA nephropathy (IgAN), membranous nephropathy (MN), and diabetic kidney disease (DKD) [12]. Polymorphisms in the genes involved in pathways such as those associated with inflammatory reactions (e.g., tumor necrosis factor (TNF)-α and interleukin (IL)-4), fibrosis (e.g., TGFB1), phase-II metabolism (e.g., GSTP1 and GSTO1), CKD worsening (e.g., UMOD) and the renin–angiotensin–aldosterone system (RAAS) (e.g., AGT and RENBP) have been suggested to affect the progression rate of patients with CKD [13].
3. Epithelial–Mesenchymal Transition (EMT)
Renal fibrosis, including nephrosclerosis and tubulointerstitial fibrosis, constitutes the final common pathway of renal injuries, regardless of etiologies. EMT is the major mechanism promoting renal fibrosis, and myofibroblasts are the main cell type that produces the extracellular matrix [14]. The origin of myofibroblasts in the kidney remains uncertain, but several candidates have been suggested, including resident fibroblasts, bone marrow-derived fibroblasts, or transition from pericytes or endothelial cells [15]. Recent research has revealed that EMT is fairly uncommon, as fibroblasts derived from EMT are rarely found in renal interstitium. A novel concept of partial EMT, indicating that tubular epithelial cells gain mesenchymal characteristics but retain their attachment to the basement membrane, may explain the pathogenic role of renal tubular epithelia in renal fibrosis [16].
After acute kidney injury (AKI) attacks, the c-Jun NH2-terminal kinase (JNK) signal is activated in tubular epithelial cells to enhance the expression of typical mesenchymal markers (e.g., e-cadherin, α-smooth muscle actin) and to upregulate profibrogenic factors (mainly transforming growth factor (TGF)-β and connective tissue growth factor (CTGF)) [17]. The persistent activation of the TGF-β pathway leading to an increased expression of SNAI1 and TWIST1 further promotes G2/M arrest. Cell cycle arrest in the G2/M phase in injured tubular cells, through JNK activation, amplifies the profibrogenic factors, such as TGF-β and CTGF, constituting a vicious cycle culminating in the fibrosis progression [18]. Fatty acid oxidation (FAO) is the main energy source of the proximal tubule (PCT). SMAD3 activated by TGF-β will suppress PPARGC1a expression to cause dysregulated FAO with lipid accumulation in the PCT, one of the characteristic features of EMT [19]. The PCT cells with lipid accumulation will enhance inflammation, innate immunity, and apoptosis to worsen renal fibrosis. Tubular cells with partial EMT can also activate fibroblasts and recruit inflammatory cells via the secretome composed of growth factors, chemokines, and cytokines, subsequently aggravating fibrosis [20]. Blocking EMT with approaches targeting the cell cycle or the inhibition of SNAI1 or TWIST1 expressions has been found to repress inflammation and fibrosis, pointing to the fact that EMT may be a good target mechanism to reverse renal fibrosis [15][18]. Bone morphogenetic protein-7 (BMP-7) can reverse EMT by counteracting the TGF-β/SMAD2/3 pathway and serves as another potential therapeutic target for improving renal injury [21]. However, the results in clinical studies of BMP-7 analogs involving patients with CKD are heterogeneous, suggesting a complex interaction between BMP-7 and other EMT-related pathways, as well as the necessity of determining the optimal serum BMP-7 concentration [22].
Epigenetic modifications, including DNA methylation and histone modification, also participate heavily in the regulation of partial EMT. The inhibition of DNA methylation was reported to ameliorate renal fibrosis. For example, low-dose hydralazine causing the de-methylation of the NASAL1 promotor and 5′-azacytidine, resulting in an inhibition of DNA methyltransferase 1 (DNMT1) [23][24][25]. Furthermore, agents targeting histone modification also confer renal benefits in CKD or AKI-to-CKD transition through the inhibition of histone methyltransferase (e.g., enhancer of zeste homolog 2) or the inhibition of histone deacetylases by directly inhibiting deacetylase (e.g., valproic acid) or indirectly interfering with histone modification readers (e.g., bromodomain and extra-terminal (BET) protein inhibitors) [26][27][28]. Epigenetics may be a novel therapeutic target for renal diseases.
4. Avoidance of Acute Kidney Injury (AKI) in Patients with CKD
AKI is associated with significant morbidity and mortality, including an increase in adverse renal outcomes. Cumulative evidence suggests that AKI is never self-limited, as it serves as a gateway to subsequent AKI episodes and, potentially, incident CKD, regardless of whether patients show recovery from AKI episodes or not [29]. Moreover, a single episode of severe AKI superimposed on patients with pre-existing CKD can cause further renal deterioration to end-stage kidney disease (ESKD) rapidly at a non-linear pace (Figure 1B). The risk factors associated with the AKI-related acceleration of renal progression were identified previously, and include older age, delayed renal function recovery from AKI, severe AKI episodes, the presence of proteinuria, and comorbidities such as DM, hypertension, and heart failure [30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][61][62][63][64][65][66][67][68][69][70][71][72][73][74][75][76][77][78][79][80][81][82][83][84][85][86][87][88][89][90][91][92][93][94][95][96][97][98][99][100][101][102][103][104][105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120][121][122][123][124][125][126][127][128][129][130][131][132]. Long-term renal sequelae become very serious if patients have acute tubular necrosis (ATN) or ischemic AKI compared with those who have other forms of AKI, suggesting that the etiology of the AKI may also be a very important risk factor [33].
Biomarkers such as kidney injury molecule (KIM)-1 and neutrophil gelatinase-associated lipocalin (NGAL) are able to detect AKI earlier than conventional indicators [34][35]. Moreover, a combination of biomarkers such as insulin-like growth factor-binding protein 7 (IGFBP7) and tissue inhibitor of metalloproteinases-2 (TIMP-2) was shown to successfully predict the development of AKI during the 12 h following blood tests and can guide the selection of interventions to prevent AKI [36][37].
The mechanisms that are responsible for the transition from AKI to CKD remain under active investigation, and a maladaptive repair response with partial epithelial–mesenchymal transition (EMT), especially at the proximal tubule (PCT), has an important role. Cell cycle arrest in the G2/M phase, the dysregulated regeneration of injured PCT due to mitochondrial dysfunction, and the aberrant activation of developmental pathways (such as the Wnt, Hedgehog, and Notch pathways) also contribute to AKI-to-CKD transition. Phenotypic changes of fibroblasts to myofibroblasts or the formation of tertiary lymphoid tissue, as well as defective switches of recruited T cells and M1 macrophages to regulatory T cells and M2 macrophages, respectively, will perpetuate inflammation and fibrosis, leading to capillary rarefaction, hypoxia, and tubule cell damage, constituting a vicious cycle [38][39][40]. Furthermore, the persistent expression of either transforming growth factor (TGF)-β or kidney injury molecule-1 (KIM-1) has also been held responsible [41][42]. Notably, several pathogenic processes contributing to AKI-to-CKD transition are also physiological repair processes within kidneys. Additional insults, including high salt and high protein intake and nephrotoxic agents, during or after AKI episodes, as well as the underlying diseased kidney (e.g., diabetic nephropathy), may turn physiological renal responses into disordered regeneration.
Identification of Factors Causing AKI
The fundamental management of AKI in patients with CKD involves preventing the occurrence of AKI, and the success of this approach largely depends on the identification of the etiologies contributing to AKI (Table 1). Furthermore, treating the underlying etiologies of AKI, the optimization of volume and hemodynamic status, the withdrawal of nephrotoxic agents, the adjustment of medication doses according to renal function, adopting a conservative (<180 mg/dL), rather than an intensive glycemic control goal, and maintaining higher mean arterial pressure in patients with underlying hypertension can be crucial strategies for AKI management [43][44]. Drug regimens should be reviewed carefully to prevent drug–drug interactions. For example, adding piperacillin/tazobactam should be avoided, as it potentiates the nephrotoxicity of vancomycin. Several agents and strategies have been tested for treating AKI, including recombinant alkaline phosphatase and L-carnitine for sepsis-related AKI, as well as p53-targeted small interfering RNA (siRNA) and remote ischemic preconditioning for surgery-associated AKI [45][46].
Table 1. Common etiologies of acute kidney injury.
| Categories | Mechanism | Examples | Evaluation |
|---|---|---|---|
| Prerenal | Cardiac output↓ | Acute myocardial infarction, valve rupture, acute pericarditis, acute myocarditis Drugs exacerbate heart failure (COX inhibitors, CCB, TZD, DPP-4i) Drugs cause direct heart injury (rheumatologic agents (e.g., TNF-α inhibitors), anthracyclines, taxanes, targeted therapy (e.g., bevacizumab, sorafenib), anti-Parkinson (Pergolide, Pramipexole) |
History: fever, vomiting, diarrhea, chest pain, orthopnea, palpitation, urine output↓, liver/CV diseases Drug: diuretics, NSAID Physical exam: BP↓/HR↑, skin turgor/mucosa, edema |
| True hypovolemia | Renal loss (diuretics, osmotic diuresis); Extrarenal loss (diarrhea, hemorrhage, burn, third spacing) | ||
| Effective volume↓ | Sepsis, neurogenic shock, anaphylaxis | ||
| Intrarenal vasoconstriction | Hypercalcemia, hepatorenal syndrome, drugs (CNIs, NSAID, vasoconstrictors.) | ||
| Intrinsic | Glomerular injury | Nephrotic (MCD, MPGN, drugs (NSAID, gold, penicillamine))213607Nephritic (IRGN, lupus nephritis, AAV, anti-GBM disease, IgAN, drugs (e.g., hydralazine)) | History: Fever, cellulitis, URI, flank pain, foamy urine, urine output↓, myalgia, hemoptysis Drug: antibiotics, NSAID, statin, contrast Physical exam: BP, Skin rash, arthritis |
| Tubular injury | Severe prerenal causes, toxins (endogenous: hemolysis, rhabdomyolysis, tumor lysis syndrome) or exogenous (aminoglycoside, contrast, CNIs, acyclovir, lithium, vancomycin)) | ||
| Interstitial injury | Allergy (drug: cephalosporin, penicillin, PPI, NSAID, herbs); Infection (bacteria, fungus, virus, leptospirosis); Autoimmune (Lupus, anti-TBM disease, AAV) | ||
| Vascular injury | Small caliber (TMA (malignant hypertension, HUS/TTP, DIC), scleroderma renal crisis) Large caliber (renal infarction, renal vein thrombosis) |
||
| Postrenal | Urinary tract | Benign prostatic hyperplasia; neurogenic bladder; Intra-ureter (stones, tumors); Extraureter (retroperitoneal fibrosis, intra-abdominal tumors) lesions | History: low urinary tract symptoms, gross hematuria Physical exam: suprapubic tenderness, abdomen mass Image: e.g., ultrasound |
| Intrarenal | Crystals (acyclovir, indinavir), stones, tumors, paraproteins (myeloma) |
Denote: AAV: ANCA associated vasculitis, COX: cyclooxygenase, CNIs: calcineurin inhibitors, DPP-4i: Dipeptidyl peptidase 4 inhibitors, DIC: disseminated intravascular coagulation, HUS: hemolytic uremic syndrome, IgAN: IgA nephropathy, MCD: minimal change disease, MPGN: membranoproliferative glomerulonephritis, NSAID: nonsteroidal anti-inflammatory drug, PPI: proton pump inhibitor, TBM: tubule basement membrane, TMA:thrombotic micrangiopathy, TNF: tumor necrosis factor, TTP: thrombotic thrombocytopenic purpura, TZD: Thiazolidinedione.
5. An Etiology-Based Treatment Strategy for CKD
As mentioned above, the different etiologies of CKD themselves have various impacts on renal progression. As an etiology-based treatment strategy for CKD has not been well addressed, we touch on this management issue for both the common and less-appreciated causes of CKD.
5.1. Glomerulopathy
Glomerulopathy is a heterogeneous group of diseases and accounts for a significant number of CKDs. It occurs more commonly in young people with non-specific presentations. Although novel diagnostic tools are under investigation, renal biopsy is still the gold standard for achieving a definite diagnosis. A slow deterioration of renal function occurs in a proportion of patients. The risk factors for a faster GFR reduction include obesity, smoking, hypertension, significant proteinuria (usually >1 g/day), CKD at the diagnosis of glomerulopathy, and pathologically chronic renal lesions (glomerulosclerosis, tubular atrophy, and interstitial fibrosis) [47]. Genetic factors contribute to a rapid GFR loss, such as APOL1 in those with FSGS [48]. Fabry disease is a frequently ignored X-link inherited disease caused by a pathogenic mutation involving GLA-encoding lysosome enzyme α-galactosidase A [49]. The deficient enzyme function results in the intracellular accumulation of globotriaosylceramide, which impairs cell metabolism. Apart from neurological and cardiovascular associations, the kidneys may also be affected by presentations of proteinuria and renal failure. Enzyme replacement therapy is the cornerstone of renal progression reduction or prevention. A brief summary of the preferred treatment for different underlying glomerulonephritis is shown in Table 2.
Table 2. The therapy of glomerulopathy and the common side effects of the pharmacotherapy.
| Non-Immune Therapy | Immunosuppressant Therapy | Denote | |||||||
|---|---|---|---|---|---|---|---|---|---|
| ↓Dietary salt/protein SGLT2 inhibitors RAAS blockades Blood pressure Infection prophylaxis (vaccine, antibioticsantiviral agents) Vitamin D + calcium |
Diuretics ↑Oncotic pressure (albumin infusion) Lipid lowering Anticoagulation (prevent or treat thromboembolism) |
Steroids | Calcineurin inhibitors | Antimetabolite | Alkalizing agents | Anti-CD20 | PP | ||
| Prednisolone ACTH |
Cyclosporine Tacrolimus |
Mycophenolate Azathioprine |
CYC Chlorambucil |
Rituximab | |||||
| MCD | ν | ν | ν | ν | ν | ν | ν | ||
| FSGS | ν | ν | ν | ν | ν | Genetic test: Congenital/infantile type, APOL1 (adult) | |||
| MN | ν | ν | ν | ν | ν | ν | ν | Serum anti-PLA2R: diagnosis, follow-up and outcomes | |
| MPGN | ν | ν | ν | ν | Treat underlying diseases (e.g., MM, lymphoma or HCV) | ||||
| IgAN | ν | ν (IgAN+MCD) | ν | ν (some RTCs) | Adjuvant antimalarial Ongoing trial: Fostamatinib, Atacicept, Bortezomib |
||||
| LN | ν | ν (class V) | ν | ν | ν | ν | Antimalarial agents AZA for maintenance |
||
| AAV | ν | ν | ν | ν | ν severe AKI PH |
Disease activity: chemokine C-X-C motif chemokine ligand 13, matrix metalloproteinase-3, tissue inhibitor of metalloproteinases-1 | |||
| Ani-GBM | ν | ν | ν | ν till anti-GBM (-) | Overlap syndrome (ANA, ANCA) | ||||
| Common S/E | Rare | Rare | ↑Glucose Cushing ↑BP |
Nephrotoxic | GI upset Leukopenia | Bone marrow suppression Infertility |
Infusion reaction, Infection Cytopenia |
Fever Urticaria | |
Denote: AAV: ANCA associated vasculitis, BP: blood pressure, CYC: cyclophosphamide, FSGS: focal segmental glomerulonephropathy, GI: gastrointestinal, GBM: glomerular basement membrane, IgAN: IgA nephropathy, LN: lupus nephritis, MCD: minimal change disease, MM: multiple myeloma, MN: membranous nephropathy, MPGN: membranoproliferative glomerulonephritis, PH: pulmonary hemorrhage, PP: plasmapheresis, RAAS: renin-agiotensin-aldosterone system, SGLT2: sodium glucose cotransporter 2, S/E: side effect.
5.2. DM-Related CKD
DM is the most common etiology of CKD and ESKD worldwide [50]. DKD usually occurs in patients with poor glycemic control, but also arises in 30–40% of patients with intensive glycemic control, suggesting a complex and multifactorial pathogenesis of DKD [51][52][53]. Clinical manifestations of DKD include impaired renal function with proteinuria. Several risk factors have been discovered, including early onset of DM, hypertension, ethnicity, obesity, the severity of proteinuria, and smoking [54][55][56][57]. In addition to adequate glycemic control, RAAS blockade is the centerpiece of DKD management. Pentoxiphylline can delay the initiation of dialysis and exert a significant antiproteinuric effect in DM patients already receiving RAAS blockades [57]. Although most statins exhibit minimal renoprotective effects [58][59][60], fenofibrate, a peroxisome proliferator-activated receptor (PPAR) α-agonist, has been shown to have an antiproteinuric effect in DKD [61]. Thiazolidinediones, a PPARγ agonist, can be beneficial for DKD, with salt and fluid retention side effects [62][63] that should be considered.
Sodium-glucose cotransporter-2 (SGLT2) inhibitors are the latest anti-hyperglycemic agent capable of decreasing blood glucose effectively by blocking glucose reabsorption in the PCT. In addition to cardiovascular benefits, SGLT2 inhibitors also postpone renal deterioration and reduce the severity of proteinuria in patients with DM (Figure 3) [64][65][66]. It is proposed that SGLT2 inhibitors protect the kidney by enhancing glycemic control, improving cardiovascular function, and decreasing body weight, as well as restoring intra and extrarenal hemodynamics, including lowering blood pressure (BP), promoting natriuresis, and re-activating tubuloglomerular feedback [67]. Structurally, SGLT2 inhibitors also are known to reverse PCT hypertrophy, which is induced by insulin resistance and hyperglycemia-related increasing sodium and glucose reabsorption through SGLT2 [68]. With the reversal of PCT hypertrophy and decreased Na+-glucose reabsorption, the kidney will be protected due to reduced energy demand and subsequently less oxidative stress, inflammation, fibrosis, and growth factor expression. Furthermore, AMPK/SIRT1 signaling, which is suppressed by hyperglycemia, could be re-activated by SGLT2 inhibitors to promote anti-inflammatory hypoxia-inducible factor (HIF)-2α and to suppress the expression of pro-inflammatory HIF-1α [69][70][71]. Hyperglycemia increases the production of reactive oxygen species (ROS), diacylglycerol (DAG), and advanced glycation end products (AGEs), all contributing to the impaired autophagic clearance of SNAI1 and activated p21 and p27. Accumulated SNAI1 and the activation of p21 and p27 result in G2/M cell cycle arrest, the hallmark of EMT, and maladaptive renal tubular regeneration. The use of SGLT2 inhibitors can restore normal autophagic clearance and inhibit pathways due to the products of hyperglycemia, attenuating cell cycle arrest-related kidney damage. Notably, it is found that the renoprotective effect may be extended to non-diabetic patients with kidney diseases [72].
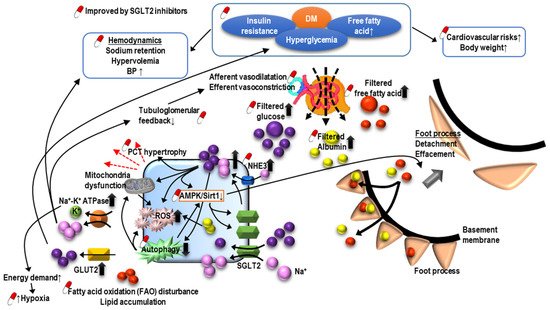
Figure 3. The pathogenesis of diabetic nephropathy and mechanism of sodium-glucose cotransporter inhibitors-associated renoprotection.
O-GlcNAcylation is a post-translational modification of proteins and can regulate various physiological or pathological processes. In DM, O-GlcNAcylation has a role in insulin resistance and can mediate glucose toxicity, and subsequently, DM complications, including DKD. Recent research has demonstrated that the O-GlcNAcylation of cellular proteins such as ICln impairs cell volume regulation in diverse cell types, a common phenomenon in DM [73]. A hypertrophic PCT is a typical feature of DKD as increased filtrated glucose promotes the reabsorption of glucose and sodium in PCT. O-GlcNAcylation may result in the cell death of PCT and contribute to the development or progression of DKD by impairing the volume regulation, which could be reversed by reducing the O-GlcNAcylation of ICln [74]. Moreover, O-GlcNAcylation in PCT was found to correlate with fatty acid oxidation [75]. These findings suggest that the manipulation of O-GlcNAcylation may be a potential target for the treatment DKD [76].
References
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 Clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150.
- Tonelli, M.; Wiebe, N.; Culleton, B.; House, A.; Rabbat, C.; Fok, M.; McAlister, F.; Garg, A.X. Chronic Kidney Disease and Mortality Risk: A Systematic Review. J. Am. Soc. Nephrol. 2006, 17, 2034–2047.
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.; O’Callaghan, C.A.; Lasserson, D.; Hobbs, R. Global Prevalence of Chronic Kidney Disease—A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0158765.
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288.
- Murton, M.; Goff-Leggett, D.; Bobrowska, A.; Garcia Sanchez, J.J.; James, G.; Wittbrodt, E.; Nolan, S.; Sörstadius, E.; Pecoits-Filho, R.; Tuttle, K. Burden of Chronic Kidney Disease by KDIGO Categories of Glomerular Filtration Rate and Albuminuria: A Systematic Review. Adv. Ther. 2021, 38, 180–200.
- Stevens, L.A.; Viswanathan, G.; Weiner, G.E. Chronic kidney disease and end-stage renal disease in the elderly population: Current prevalence, future projections, and clinical significance. Adv. Chronic Kidney Dis. 2010, 17, 293–301.
- Liu, M.; Liu, S.-W.; Wang, L.-J.; Bai, Y.-M.; Zeng, X.-Y.; Guo, H.-B.; Liu, Y.-N.; Jiang, Y.-Y.; Dong, W.-L.; He, G.-X.; et al. Burden of diabetes, hyperglycaemia in China from to 2016: Findings from the 1990 to 2016, global burden of disease study. Diabetes Metab. 2018, 45, 286–293.
- Duan, J.; Wang, C.; Liu, D.; Qiao, Y.; Pan, S.; Jiang, D.; Zhao, Z.; Liang, L.; Tian, F.; Yu, P.; et al. Prevalence and risk factors of chronic kidney disease and diabetic kidney disease in Chinese rural residents: A cross-sectional survey. Sci. Rep. 2019, 9, 10408.
- Li, L.; Astor, B.C.; Lewis, J.; Hu, B.; Appel, L.J.; Lipkowitz, M.S.; Toto, R.D.; Wang, X.; Wright, J.T.; Greene, T.H. Longitudinal Progression Trajectory of GFR Among Patients With CKD. Am. J. Kidney Dis. 2012, 59, 504–512.
- Lee, D.E.; Qamar, M.; Wilke, R.A. Relative Contribution of Genetic and Environmental Factors in CKD. S. Dak. Med. 2021, 74, 306–309.
- Beyer-Westendorf, J.; Kreutz, R.; Posch, F.; Ay, C. The CHA2DS2-VASc score strongly correlates with glomerular filtration rate and predicts renal function decline over time in elderly patients with atrial fibrillation and chronic kidney disease. Int. J. Cardiol. 2018, 253, 71–77.
- Remuzzi, G.; Benigni, A.; Remuzzi, A. Mechanisms of progression and regression of renal lesions of chronic nephropathies and diabetes. J. Clin. Investig. 2006, 116, 288–296.
- Cañadas-Garre, M.; Anderson, K.; Cappa, R.; Skelly, R.; Smyth, L.J.; McKnight, A.J.; Maxwell, A.P. Genetic Susceptibility to Chronic Kidney Disease—Some More Pieces for the Heritability Puzzle. Front. Genet. 2019, 10, 453.
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36.
- Grande, M.T.; Sanchez-Laorden, B.; Lopez-Blau, C.; De Frutos, C.A.; Boutet, A.; Arevalo, M.; Rowe, R.G.; Weiss, S.J.; Lopez-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to re-verse established disease. Nat. Med. 2015, 21, 989–997.
- Hajarnis, S.; Yheskel, M.; Williams, D.; Brefort, T.; Glaudemans, B.; Debaix, H.; Baum, M.; Devuyst, O.; Patel, V. Suppression of microRNA Activity in Kidney Collecting Ducts Induces Partial Loss of Epithelial Phenotype and Renal Fibrosis. J. Am. Soc. Nephrol. 2018, 29, 518–531.
- Canaud, G.; Bonventre, V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transpl. 2015, 30, 575–583.
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.-C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009.
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPAR α and the Fatty Acid Oxidation Pathway Ag-gravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237.
- Liu, B.-C.; Tang, T.-T.; Lv, L.-L.; Lan, H.-Y. Renal tubule injury: A driving force toward chronic kidney disease. Kidney Int. 2018, 93, 568–579.
- Song, Y.; Lv, S.; Wang, F.; Liu, X.; Cheng, J.; Liu, S.; Wang, X.; Chen, W.; Guan, G.; Liu, G.; et al. Overexpression of BMP-7 reverses TGF-β1-induced epithelial-mesenchymal transition. Mol. Med. Rep. 2020, 21, 833–841.
- Tampe, B.; Tampe, D.; Nyamsuren, G.; Klöpper, F.; Rapp, G.; Kauffels, A.; Lorf, T.; Zeisberg, E.M.; Müller, G.A.; Kalluri, R.; et al. Pharmacological induction of hypoxia-inducible transcription factor ARNT attenuates chronic kidney failure. J. Clin. Investig. 2018, 128, 3053–3070.
- Tampe, B.; Tampe, D.; Zeisberg, E.M.; Müller, G.A.; Bechtel-Walz, W.; Koziolek, M.; Kalluri, R.; Zeisberg, M. Induction of Tet3-dependent Epigenetic Remodeling by Low-dose Hydralazine Attenuates Progression of Chronic Kidney Disease. EBioMedicine 2014, 2, 19–36.
- Tampe, B.; Steinle, U.; Tampe, D.; Carstens, J.; Korsten, P.; Zeisberg, E.M.; Müller, G.A.; Kalluri, R.; Zeisberg, M. Low-dose hydralazine prevents fibrosis in a murine model of acute kidney injury–to–chronic kidney disease progression. Kidney Int. 2016, 91, 157–176.
- Larkin, B.P.; Saad, S.; Glastras, S.J.; Nguyen, L.T.; Hou, M.; Chen, H.; Wang, R.; Pollock, C.A. Low-dose hydralazine during gestation reduces renal fibrosis in rodent offspring exposed to maternal high fat diet. PLoS ONE 2021, 16, e0248854.
- Zhou, X.; Zang, X.; Ponnusamy, M.; Masucci, M.V.; Tolbert, E.; Gong, R.; Zhao, T.C.; Liu, N.; Bayliss, G.; Dworkin, L.D.; et al. Enhancer of Zeste Homolog 2 Inhibition Attenuates Renal Fibrosis by Maintaining Smad7 and Phosphatase and Tensin Homolog Ex-pression. J. Am. Soc. Nephrol. 2016, 27, 2092–2108.
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Pro-teins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315.
- Mello, M.L.S. Sodium Valproate-Induced Chromatin Remodeling. Front. Cell Dev. Biol. 2021, 9, 645518.
- Heung, M.; Steffick, D.E.; Zivin, K.; Gillespie, B.W.; Banerjee, T.; Hsu, C.-Y.; Powe, N.R.; Pavkov, M.E.; Williams, D.E.; Saran, R.; et al. Acute Kidney Injury Recovery Pattern and Subsequent Risk of CKD: An Analysis of Veterans Health Administration Data. Am. J. Kidney Dis. 2016, 67, 742–752.
- Ishani, A.; Xue, J.L.; Himmelfarb, J.; Eggers, P.W.; Kimmel, P.L.; Molitoris, B.A.; Collins, A.J. Acute Kidney Injury Increases Risk of ESRD among Elderly. J. Am. Soc. Nephrol. 2008, 20, 223–228.
- Liu, K.D.; Yang, J.; Tan, T.C.; Glidden, D.V.; Zheng, S.; Pravoverov, L.; Hsu, C.Y.; Go, A.S. Risk Factors for Recurrent Acute Kidney Injury in a Large Population-Based Cohort. Am. J. Kidney Dis. 2019, 73, 163–173.
- Coca, S.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448.
- Amdur, R.L.; Chawla, L.S.; Amodeo, S.; Kimmel, P.L.; Palant, C.E. Outcomes following diagnosis of acute renal failure in U.S. veterans: Focus on acute tubular necrosis. Kidney Int. 2009, 76, 1089–1097.
- Kashani, K.; Kellum, J.A. Novel biomarkers indicating repair or progression after acute kidney injury. Curr. Opin. Nephrol. Hypertens. 2015, 24, 21–27.
- Sabbisetti, V.S.; Waikar, S.S.; Antoine, D.J.; Smiles, A.; Wang, C.; Ravisankar, A.; Ito, K.; Sharma, S.; Ramadesikan, S.; Lee, M.; et al. Blood Kidney Injury Molecule-1 Is a Biomarker of Acute and Chronic Kidney Injury and Predicts Progression to ESRD in Type I Diabetes. J. Am. Soc. Nephrol. 2014, 25, 2177–2186.
- Teo, S.H.; Endre, Z.H. Biomarkers in acute kidney injury (AKI). Best Pract. Res. Clin. Anaesthesiol. 2017, 31, 331–344.
- Göcze, I.; Jauch, D.; Götz, M.; Kennedy, P.; Jung, B.; Zeman, F.; Gnewuch, C.; Graf, B.M.; Gnann, W.; Banas, B.; et al. Biomarker-guided Intervention to Prevent Acute Kidney Injury After Major Surgery: The Prospective Randomized BigpAK Study. Ann. Surg. 2018, 267, 1013–1020.
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543.
- Basile, D. The endothelial cell in ischemic acute kidney injury: Implications for acute and chronic function. Kidney Int. 2007, 72, 151–156.
- Little, M.H.; Kairath, P. Does Renal Repair Recapitulate Kidney Development? J. Am. Soc. Nephrol. 2017, 28, 34–46.
- Kramann, R.; Wongboonsin, J.; Chang-Panesso, M.; Machado, F.G.; Humphreys, B.D. Gli1 + Pericyte Loss Induces Capillary Rare-faction and Proximal Tubular Injury. J. Am. Soc. Nephrol. 2017, 28, 776–784.
- Sato, Y.; Yanagita, M. Immune cells and inflammation in AKI to CKD progression. Am. J. Physiol. Physiol. 2018, 315, F1501–F1512.
- NICE-SUGAR Study Investigators; Finfer, S.; Chittock, D.R.; Su, S.Y.; Blair, D.; Foster, D.; Dhingra, V.; Bellomo, R.; Cook, D.; Dodek, P.; et al. Intensive versus conventional glucose control in critically ill patients. N. Engl. J. Med. 2009, 360, 1283–1297.
- Asfar, P.; Meziani, F.; Hamel, J.-F.; Grelon, F.; Mégarbane, B.; Anguel, N.; Mira, J.-P.; Dequin, P.-F.; Gergaud, S.; Weiss, N.; et al. High versus Low Blood-Pressure Target in Patients with Septic Shock. N. Engl. J. Med. 2014, 370, 1583–1593.
- Kaushal, G.P.; Shah, S.V. Challenges and Advances in the Treatment of AKI. J. Am. Soc. Nephrol. 2014, 25, 877–883.
- Hausenloy, D.J.; Candilio, L.; Evans, R.; Ariti, C.; Jenkins, D.P.; Kolvekar, S.; Knight, R.; Kunst, G.; Laing, C.; Nicholas, J.; et al. Remote Ischemic Preconditioning and Outcomes of Cardiac Surgery. N. Engl. J. Med. 2015, 373, 1408–1417.
- Floege, J.; Amann, K. Primary glomerulonephritides. Lancet 2016, 387, 2036–2048.
- Friedman, D.; Pollak, M.R. APOL1and Kidney Disease: From Genetics to Biology. Annu. Rev. Physiol. 2020, 82, 323–342.
- Turkmen, K.; Baloglu, I. Fabry disease: Where are we now? Int. Urol. Nephrol. 2020, 52, 2113–2122.
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252.
- Gnudi, L.; Coward, R.M.; Long, D.A. Diabetic Nephropathy: Perspective on Novel Molecular Mechanisms. Trends Endocrinol. Metab. 2016, 27, 820–830.
- Vallon, V.; Gerasimova, M.; Rose, M.A.; Masuda, T.; Satriano, J.; Mayoux, E.; Koepsell, H.; Thomson, S.C.; Rieg, T. SGLT2 inhibitor em-pagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am. J. Physiol. Renal. Physiol. 2014, 306, F194–F204.
- Tonneijck, L.; Muskiet, M.H.A.; Smits, M.M.; van Bommel, E.J.; Heerspink, H.J.L.; van Raalte, D.H.; Joles, J.A. Glomerular Hyperfiltration in Diabetes: Mechanisms, Clinical Significance, and Treatment. J. Am. Soc. Nephrol. 2017, 28, 1023–1039.
- Thomas, M.C.; Brownlee, M.; Susztak, K.; Sharma, K.; Jandeleit-Dahm, K.A.; Zoungas, S.; Rossing, P.; Groop, P.H.; Cooper, M.E. Diabetic kidney disease. Nat. Rev. Dis. Primers 2015, 1, 15018.
- Ravid, M.; Brosh, D.; Ravid-Safran, D.; Levy, Z.; Rachmani, R. Main Risk Factors for Nephropathy in Type 2 Diabetes Mellitus Are Plasma Cholesterol Levels, Mean Blood Pressure, and Hyperglycemia. Arch. Intern. Med. 1998, 158, 998–1004.
- Alicic, R.Z.; Rooney, M.T.; Tuttle, K.R. Diabetic Kidney Disease: Challenges, Progress, and Possibilities. Clin. J. Am. Soc. Nephrol. 2017, 12, 2032–2045.
- Navarro-González, J.F.; Mora-Fernández, C.; Muros de Fuentes, M.; Chahin, J.; Méndez, M.L.; Gallego, E.; Macía, M.; del Castillo, N.; Rivero, A.; Getino, M.A.; et al. Effect of Pentoxifylline on Renal Function and Urinary Albumin Excretion in Patients with Diabetic Kidney Disease: The PREDIAN Trial. J. Am. Soc. Nephrol. 2015, 26, 220–229.
- Vogt, L.; Bangalore, S.; Fayyad, R.; Melamed, S.; Hovingh, G.K.; DeMicco, D.A.; Waters, D.D. Atorvastatin Has a Dose-Dependent Beneficial Effect on Kidney Function and Associated Cardiovascular Outcomes: Post Hoc Analysis of 6 Double-Blind Ran-domized Controlled Trials. J. Am. Heart Assoc. 2019, 8, e010827.
- De Zeeuw, D.; Anzalone, D.A.; Cain, V.A.; Cressman, M.D.; Heerspink, H.J.; Molitoris, B.A.; Monyak, J.T.; Parving, H.H.; Remuzzi, G.; Sowers, J.R.; et al. Renal effects of atorvastatin and rosuvastatin in patients with diabetes who have progressive renal disease (PLANET I): A randomised clinical trial. Lancet Diabetes Endocrinol. 2015, 3, 181–190.
- Rutter, M.; Prais, H.R.; Charlton-Menys, V.; Gittins, M.; Roberts, C.; Davies, R.R.; Moorhouse, A.; Jinadev, P.; France, M.; Wiles, P.G.; et al. Protection Against Nephropathy in Diabetes with Atorvastatin (PANDA): A randomized double-blind placebo-controlled trial of high- vs. low-dose atorvastatin1. Diabet. Med. 2010, 28, 100–108.
- Ansquer, J.C.; Foucher, C.; Rattier, S.; Taskinen, M.R.; Steiner, G.; DAIS Investigators. Fenofibrate reduces progression to microalbu-minuria over 3 years in a placebo-controlled study in type 2 diabetes: Results from the Diabetes Atherosclerosis Intervention Study (DAIS). Am. J. Kidney Dis. 2005, 45, 485–493.
- Miyazaki, Y.; Cersosimo, E.; Triplitt, C.; DeFronzo, R. Rosiglitazone decreases albuminuria in type 2 diabetic patients. Kidney Int. 2007, 72, 1367–1373.
- Sarafidis, P.A. Thiazolidinediones and diabetic nephropathy: Need for a closer examination? J. Cardiometab. Syndr. Fall 2007, 2, 297–301.
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; De Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657.
- Wiviott, S.D.; Raz, I.; Bonaca, M.P.; Mosenzon, O.; Kato, E.; Cahn, A.; Trimarco, B.; Zelniker, T.A.; Kuder, J.F.; Murphy, S.A.; et al. Dapagliflozin and Cardiovascular Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2019, 380, 347–357.
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128.
- Alicic, R.Z.; Neumiller, J.J.; Johnson, E.J.; Dieter, B.; Tuttle, K. Sodium–Glucose Cotransporter 2 Inhibition and Diabetic Kidney Disease. Diabetes 2019, 68, 248–257.
- Tejedor Jorge, A. Hemodynamic and renal implications of sodium-glucose cotransporter- 2 inhibitors in type 2 diabetes mellitus. Med. Clin. 2016, 147, 35–43.
- Chilton, R.; Tikkanen, I.; Cannon, C.P.; Crowe, S.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 1180–1193.
- Heerspink, H.J.; Perkins, B.A.; Fitchett, D.H.; Husain, M.; Cherney, D.Z. Sodium Glucose Cotransporter 2 Inhibitors in the Treatment of Diabetes Mellitus: Cardiovascular and Kidney Effects, Potential Mechanisms, and Clinical Applications. Circulation 2016, 134, 752–772.
- Kalra, S. Sodium Glucose Co-Transporter-2 (SGLT2) Inhibitors: A Review of Their Basic and Clinical Pharmacology. Diabetes Ther. 2014, 5, 355–366.
- Fernandez-Fernandez, B.; Sarafidis, P.; Kanbay, M.; Navarro-González, J.F.; Soler, M.J.; Górriz, J.L.; Ortiz, A. SGLT2 inhibitors for non-diabetic kidney disease: Drugs to treat CKD that also improve glycaemia. Clin. Kidney J. 2020, 13, 728–733.
- Costa, R.; Remigante, A.; Civello, D.A.; Bernardinelli, E.; Szabó, Z.; Morabito, R.; Marino, A.; Sarikas, A.; Patsch, W.; Paulmichl, M.; et al. O-GlcNAcylation Suppresses the Ion Current IClswell by Preventing the Binding of the Protein ICln to α-Integrin. Front. Cell Dev. Biol. 2020, 8, 607080.
- Okada, Y.; Numata, T.; Sato-Numata, K.; Sabirov, R.Z.; Liu, H.; Mori, S.I.; Morishima, S. Roles of volume-regulatory anion channels, VSOR and Maxi-Cl, in apoptosis, cisplatin resistance, necrosis, ischemic cell death, stroke and myocardial infarction. Curr. Top. Membr. 2019, 83, 205–283.
- Sugahara, S.; Kume, S.; Chin-Kanasaki, M.; Tomita, I.; Yasuda-Yamahara, M.; Yamahara, K.; Takeda, N.; Osawa, N.; Yanagita, M.; Araki, S.I.; et al. Protein O-GlcNAcylation Is Essential for the Maintenance of Renal Energy Homeostasis and Function via Lipolysis during Fasting and Diabetes. J. Am. Soc. Nephrol. 2019, 30, 962–978.
- Furst, J.; Gschwentner, M.; Ritter, M.; Botta, G.; Jakab, M.; Mayer, M.; Garavaglia, L.; Bazzini, C.; Rodighiero, S.; Meyer, G.; et al. Molecular and functional aspects of anionic channels activated during regulatory volume decrease in mammalian cells. Pflugers Arch. 2002, 444, 1–25.
- Ku, E.; Lee, B.J.; Wei, J.; Weir, M.R. Hypertension in CKD: Core Curriculum 2019. Am. J. Kidney Dis. 2019, 74, 120–131.
- Taler, S.J.; Agarwal, R.; Bakris, G.L.; Flynn, J.T.; Nilsson, P.M.; Rahman, M.; Sanders, P.; Textor, S.C.; Weir, M.R.; Townsend, R.R. KDOQI US Commentary on the 2012 KDIGO Clinical Practice Guideline for Management of Blood Pressure in CKD. Am. J. Kidney Dis. 2013, 62, 201–213.
- Kidney Disease: Improving Global Outcomes (KDIGO) Blood Pressure Work Group. KDIGO 2021 Clinical Practice Guideline for the Management of Blood Pressure in Chronic Kidney Disease. Kidney Int 2021, 99, S1–S87.
- Hsu, C.-N.; Tain, Y.-L. Gasotransmitters for the Therapeutic Prevention of Hypertension and Kidney Disease. Int. J. Mol. Sci. 2021, 22, 7808.
- Bakris, G.; Hart, P.; Ritz, E. Beta blockers in the management of chronic kidney disease. Kidney Int. 2006, 70, 1905–1913.
- Pugh, D.; Gallacher, P.J.; Dhaun, N. Management of Hypertension in Chronic Kidney Disease. Drugs 2019, 79, 365–379.
- Chung, E.Y.; Ruospo, M.; Natale, P.; Bolignano, D.; Navaneethan, S.D.; Palmer, S.C.; Strippoli, G.F. Aldosterone antagonists in addition to renin angiotensin system antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst. Rev. 2020, 10, CD007004.
- Bolignano, D.; Palmer, S.C.; Navaneethan, S.D.; Strippoli, G.F. Aldosterone antagonists for preventing the progression of chronic kidney disease. Cochrane Database Syst. Rev. 2014, 29, CD007004.
- Takahashi, S.; Katada, J.; Daida, H.; Kitamura, F.; Yokoyama, K. Effects of mineralocorticoid receptor antagonists in proteinuric kidney disease: A systematic review and meta-analysis of randomized controlled trials. J. Hypertens. 2019, 37, 2307–2324.
- Grune, J.; Beyhoff, N.; Smeir, E.; Chudek, R.; Blumrich, A.; Bán, Z.; Brix, S.; Betz, I.R.; Schupp, M.; Foryst-Ludwig, A.; et al. Selective Mineralocorticoid Receptor Cofactor Modulation as Molecular Basis for Finerenone’s Antifibrotic Activity. Hypertension 2018, 71, 599–608.
- Agarwal, R.; Kolkhof, P.; Bakris, G.; Bauersachs, J.; Haller, H.; Wada, T.; Zannad, F. Steroidal and non-steroidal mineralocorticoid receptor antagonists in cardiorenal medicine. Eur Heart J. 2021, 42, 152–161.
- Pitt, B.; Kober, L.; Ponikowski, P.; Gheorghiade, M.; Filippatos, G.; Krum, H.; Nowack, C.; Kolkhof, P.; Kim, S.-Y.; Zannad, F. Safety and tolerability of the novel non-steroidal mineralocorticoid receptor antagonist BAY 94-8862 in patients with chronic heart failure and mild or moderate chronic kidney disease: A randomized, double-blind trial. Eur. Hear. J. 2013, 34, 2453–2463.
- Bakris, G.L.; Agarwal, R.; Anker, S.D.; Pitt, B.; Ruilope, L.M.; Rossing, P.; Kolkhof, P.; Nowack, C.; Schloemer, P.; Joseph, A.; et al. Effect of Finerenone on Chronic Kidney Disease Outcomes in Type 2 Diabetes. N. Engl. J. Med. 2020, 383, 2219–2229.
- Ito, S.; Kashihara, N.; Shikata, K.; Nangaku, M.; Wada, T.; Okuda, Y.; Sawanobori, T. Esaxerenone (CS-3150) in Patients with Type 2 Diabetes and Microalbuminuria (ESAX-DN): Phase 3 Randomized Controlled Clinical Trial. Clin. J. Am. Soc. Nephrol. 2020, 15, 1715–1727.
- Itoh, H.; Ito, S.; Rakugi, H.; Okuda, Y.; Nishioka, S. Efficacy and safety of dosage-escalation of low-dosage esaxerenone added to a RAS inhibitor in hypertensive patients with type 2 diabetes and albuminuria: A single-arm, open-label study. Hypertens. Res. 2019, 42, 1572–1581.
- Rico-Mesa, J.S.; White, A.; Ahmadian-Tehrani, A.; Anderson, A.S. Mineralocorticoid Receptor Antagonists: A Comprehensive Re-view of Finerenone. Curr. Cardiol. Rep. 2020, 22, 140.
- Solomon, S.D.; Vaduganathan, M.; Claggett, B.L.; Packer, M.; Zile, M.; Swedberg, K.; Rouleau, J.; Pfeffer, M.A.; Desai, A.; Lund, L.H.; et al. Sacubitril/Valsartan Across the Spectrum of Ejection Fraction in Heart Failure. Circulation 2020, 141, 352–361.
- Packer, M.; Claggett, B.; Lefkowitz, M.P.; McMurray, J.J.V.; Rouleau, J.L.; Solomon, S.D.; Zile, M.R. Effect of neprilysin inhibition on renal function in patients with type 2 diabetes and chronic heart failure who are receiving target doses of inhibitors of the ren-in-angiotensin system: A secondary analysis of the PARADIGM-HF trial. Lancet Diab. Endocrinol. 2018, 6, 547–554.
- Collins, A.J.; Foley, R.N.; Herzog, C.; Chavers, B.; Gilbertson, D.; Ishani, A.; Kasiske, B.; Liu, J.; Mau, L.W.; McBean, M.; et al. US Renal Data System 2010 Annual Data Report. Am. J. Kidney Dis. 2011, 57, A8.
- Gillen, D.L.; Worcester, E.M.; Coe, F.L. Decreased renal function among adults with a history of nephrolithiasis: A study of NHANES III. Kidney Int. 2005, 67, 685–690.
- Uribarri, J. Chronic kidney disease and kidney stones. Curr. Opin. Nephrol. Hypertens. 2020, 29, 237–242.
- Tang, X.; Lieske, J.C. Acute and chronic kidney injury in nephrolithiasis. Curr. Opin. Nephrol. Hypertens. 2014, 23, 385–390.
- Rule, A.D.; Krambeck, A.E.; Lieske, J.C. Chronic Kidney Disease in Kidney Stone Formers. Clin. J. Am. Soc. Nephrol. 2011, 6, 2069–2075.
- Gambaro, G.; Favaro, S.; D’Angelo, A. Risk for Renal Failure in Nephrolithiasis. Am. J. Kidney Dis. 2001, 37, 233–243.
- Hoppe, B.; Martin-Higueras, C. Inherited conditions resulting in nephrolithiasis. Curr. Opin. Pediatr. 2020, 32, 273–283.
- Edvardsson, V.O.; Goldfarb, D.; Lieske, J.C.; Lasic, L.B.; Anglani, F.; Milliner, D.S.; Palsson, R. Hereditary causes of kidney stones and chronic kidney disease. Pediatr. Nephrol. 2013, 28, 1923–1942.
- Tran, T.V.M.; Maalouf, N.M. Uric acid stone disease: Lessons from recent human physiologic studies. Curr. Opin. Nephrol. Hypertens. 2020, 29, 407–413.
- Garrelfs, S.F.; Frishberg, Y.; Hulton, S.A.; Koren, M.J.; O’Riordan, W.D.; Cochat, P.; Deschênes, G.; Shasha-Lavsky, H.; Saland, J.M.; Hoff, W.G.V.; et al. Lumasiran, an RNAi Therapeutic for Primary Hyperoxaluria Type 1. N. Engl. J. Med. 2021, 384, 1216–1226.
- Chapman, A.B.; Devuyst, O.; Eckardt, K.-U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27.
- Levy, M.; Feingold, J. Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 2000, 58, 925–943.
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207.
- Cornec-Le Gall, E.; Alam, A.; Perrone, R.D. Autosomal dominant polycystic kidney disease. Lancet 2019, 393, 919–935.
- Kim, D.Y.; Park, J.H. Genetic Mechanisms of ADPKD. Cystogenesis 2016, 933, 13–22.
- Lanktree, M.; Chapman, A.B. New treatment paradigms for ADPKD: Moving towards precision medicine. Nat. Rev. Nephrol. 2017, 13, 750–768.
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Dandurand, A.; Ouyang, J.; Czerwiec, F.S.; Blais, J.D.; for the TEMPO 4:4 Trial Investigators. Multicenter, open-label, extension trial to evaluate the long-term efficacy and safety of early versus delayed treatment with tolvaptan in autosomal dominant polycystic kidney disease: The TEMPO 4:4 Trial. Nephrol. Dial. Transplant. 2017, 33, 477–489.
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S. Tolvaptan in Patients with Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2012, 367, 2407–2418.
- Bleyer, A.J.; Kidd, K.; Živná, M.; Kmoch, S. Autosomal Dominant Tubulointerstitial Kidney Disease. Adv. Chronic Kidney Dis. 2017, 24, 86–93.
- Devuyst, O.; Olinger, E.; Weber, S.; Eckardt, K.U.; Kmoch, S.; Rampoldi, L.; Bleyer, A.J. Autosomal dominant tubulointerstitial kidney disease. Nat. Rev. Dis. Primers 2019, 5, 60.
- Eckardt, K.-U.; Alper, S.L.; Antignac, C.; Bleyer, A.J.; Chauveau, D.; Dahan, K.; Deltas, C.; Hosking, A.; Kmoch, S.; Rampoldi, L.; et al. Autosomal dominant tubulointerstitial kidney disease: Diagnosis, classification, and management—A KDIGO consensus report. Kidney Int. 2015, 88, 676–683.
- Lim, M.A.; Kohli, J.; Bloom, R.D. Immunosuppression for kidney transplantation: Where are we now and where are we going? Transplant. Rev. 2017, 31, 10–17.
- De Lucena, D.D.; Rangel, É.B. Glucocorticoids use in kidney transplant setting. Expert Opin. Drug Metab. Toxicol. 2018, 14, 1023–1041.
- Clayton, P.A.; McDonald, S.P.; Chapman, J.R.; Chadban, S.J. Mycophenolate versus azathioprine for kidney transplantation: A 15-year follow-up of a randomized trial. Transplantation 2012, 94, 152–158.
- Casey, M.J.; Meier-Kriesche, H.U. Calcineurin inhibitors in kidney transplantation: Friend or foe? Curr. Opin. Nephrol. Hypertens. 2011, 20, 610–615.
- Vanhove, T.; Annaert, P.; Kuypers, D.R. Clinical determinants of calcineurin inhibitor disposition: A mechanistic review. Drug Metab. Rev. 2016, 48, 88–112.
- Fantus, D.; Rogers, N.M.; Grahammer, F.; Huber, T.B.; Thomson, A.W. Roles of mTOR complexes in the kidney: Implications for renal disease and transplantation. Nat. Rev. Nephrol. 2016, 12, 587–609.
- Noble, J.; Jouve, T.; Janbon, B.; Rostaing, L.; Malvezzi, P. Belatacept in kidney transplantation and its limitations. Expert Rev. Clin. Immunol. 2019, 15, 359–367.
- Wong, W.; Venetz, J.-P.; Tolkoff-Rubin, N.; Pascual, M. 2005 Immunosuppressive Strategies in Kidney Transplantation: Which Role for the Calcineurin Inhibitors? Transplantation 2005, 80, 289–296.
- Tedesco-Silva, H.; Rial, M.D.C.; Santiago, J.C.; Mazzali, M.; Pacheco-Silva, A.; Torres, R. Optimizing the Clinical Utility of Sirolimus-Based Immunosuppression for Kidney Transplantation. Clin. Transplant. 2018, 33, e13464.
- Préterre, J.; Visentin, J.; Saint Cricq, M.; Kaminski, H.; Del Bello, A.; Prezelin-Reydit, M.; Merville, P.; Kamar, N.; Couzi, L. Comparison of two strategies based on mammalian target of rapamycin inhibitors in secondary prevention of non-melanoma skin cancer after kidney transplantation, a pilot study. Clin. Transplant. 2021, 35, e14207.
- Kuczera, P.; Ciaston-Mogilska, D.; Oslizlo, B.; Hycki, A.; Wiecek, A.; Adamczak, M. The Prevalence of Metabolic Acidosis in Patients with Different Stages of Chronic Kidney Disease: Single-Centre Study Kidney. Blood Press Res. 2020, 45, 863–872.
- Di Iorio, B.R.; Bellasi, A.; Raphael, K.L.; Santoro, D.; Aucella, F.; Garofano, L.; Ceccarelli, M.; Di Lullo, L.; Capolongo, G.; Di Iorio, M.; et al. Treatment of metabolic acidosis with sodium bicarbonate delays progression of chronic kidney disease: The UBI Study. J. Nephrol. 2019, 32, 989–1001.
- Nagami, G.T. Role of angiotensin II in the enhancement of ammonia production and secretion by the proximal tubule in met-abolic acidosis. Am. J. Physiol. Renal Physiol. 2008, 294, 874–880.
- Wesson, D.E.; Simoni, J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to pro-gressive GFR decline, a situation ameliorated by alkali diet. Kidney Int. 2010, 78, 1128–1135.
- Wesson, D.E.; Nathan, T.; Rose, T.; Simoni, J.; Tran, R.M. Dietary protein induces endothelin-mediated kidney injury through en-hanced intrinsic acid production. Kidney Int. 2007, 71, 210–217.
- Kovesdy, C.P.; Anderson, J.E.; Kalantar-Zadeh, K. Association of serum bicarbonate levels with mortality in patients with non-dialysis-dependent CKD. Nephrol. Dial. Transplant. 2008, 24, 1232–1237.
- Levy, B.; Collin, S.; Sennoun, N.; Ducrocq, N.; Kimmoun, A.; Asfar, P.; Perez, P.; Meziani, F. Vascular hyporesponsiveness to vaso-pressors in septic shock: From bench to bedside. Intensive Care Med. 2010, 36, 2019–2029.
More
Information
Subjects:
Urology & Nephrology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.2K
Revisions:
2 times
(View History)
Update Date:
08 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No