 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simone Allegrini | + 2736 word(s) | 2736 | 2021-08-17 10:10:51 | | | |
| 2 | Vivi Li | Meta information modification | 2736 | 2021-09-29 11:18:25 | | |
Video Upload Options
The enzymes of both de novo and salvage pathways for purine nucleotide synthesis are regulated to meet the demand of nucleic acid precursors during proliferation. Among them, the salvage pathway enzymes seem to play the key role in replenishing the purine pool in dividing and tumour cells that require a greater amount of nucleotides. An imbalance in the purine pools is fundamental not only for preventing cell proliferation, but also, in many cases, to promote apoptosis. It is known that tumour cells harbour several mutations that might lead to defective apoptosis-inducing pathways, and this is probably at the basis of the initial expansion of the population of neoplastic cells. Therefore, knowledge of the molecular mechanisms that lead to apoptosis of tumoural cells is key to predicting the possible success of a drug treatment and planning more effective and focused therapies.
1. Introduction
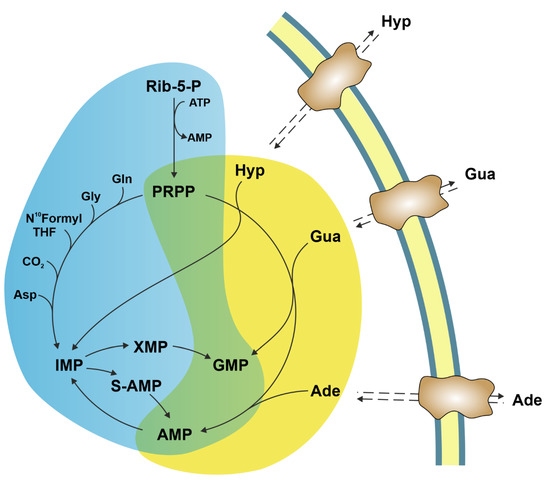
2. Ectosolic 5′-Nucleotidase
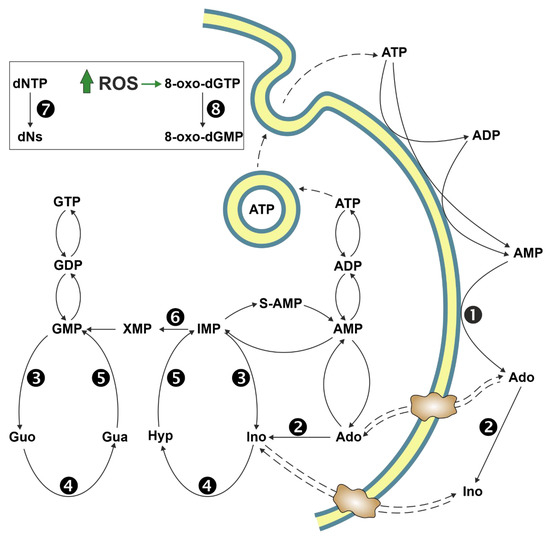
3. Cytosolic 5′-Nucleotidase II
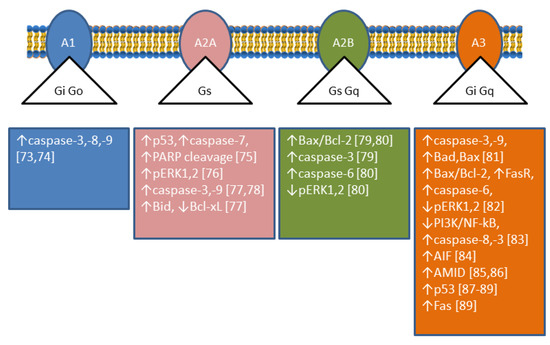
4. Adenosine Deaminase
References
- Ipata, P.L.; Balestri, F.; Camici, M.; Tozzi, M.G. Molecular mechanisms of nucleoside recycling in the brain. Int. J. Biochem. Cell Biol. 2011, 43, 140–145.
- An, S.; Kumar, R.; Sheets, E.D.; Benkovic, S.J. Reversible compartmentalization of de novo purine biosynthetic complexes in living cells. Science 2008, 320, 103–106.
- Pedley, A.M.; Benkovic, S.J. A new view into the regulation of purine metabolism: The purinosome. Trends Biochem. Sci. 2017, 42, 141–154.
- Rampazzo, C.; Miazzi, C.; Franzolin, E.; Pontarin, G.; Ferraro, P.; Frangini, M.; Reichard, P.; Bianchi, V. Regulation by degradation, a cellular defense against deoxyribonucleotide pool imbalances. Mutat. Res. 2010, 703, 2–10.
- Zhang, B. CD73 promotes tumor growth and metastasis. Oncoimmunology 2012, 1, 67–70.
- Gao, Z.W.; Dong, K.; Zhang, H.Z. The roles of CD73 in cancer. Biomed. Res. Int. 2014, 2014, 460654.
- Regateiro, F.S.; Cobbold, S.P.; Waldmann, H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin. Exp. Immunol. 2013, 171, 1–7.
- Kazemi, M.H.; Raoofi Mohseni, S.; Hojjat-Farsangi, M.; Anvari, E.; Ghalamfarsa, G.; Mohammadi, H.; Jadidi-Niaragh, F. Adenosine and adenosine receptors in the immunopathogenesis and treatment of cancer. J. Cell Physiol. 2018, 233, 2032–2057.
- Sousa, J.B.; Fresco, P.; Diniz, C.; Goncalves, J. Adenosine receptor ligands on cancer therapy: A review of patent literature. Recent Pat. Anticancer Drug Discov. 2018, 13, 40–69.
- Sadej, R.; Skladanowski, A.C. Dual, enzymatic and non-enzymatic, function of ecto-5’-nucleotidase (eN, CD73) in migration and invasion of A375 melanoma cells. Acta Biochim. Pol. 2012, 59, 647–652.
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552.
- Yang, X.; Pei, S.; Wang, H.; Jin, Y.; Yu, F.; Zhou, B.; Zhang, H.; Zhang, D.; Lin, D. Tiamulin inhibits breast cancer growth and pulmonary metastasis by decreasing the activity of CD73. BMC Cancer 2017, 17, 255.
- Yu, J.; Wang, X.; Lu, Q.; Wang, J.; Li, L.; Liao, X.; Zhu, W.; Lv, L.; Zhi, X.; Yu, J.; et al. Extracellular 5’-nucleotidase (CD73) promotes human breast cancer cells growth through AKT/GSK-3beta/beta-catenin/cyclinD1 signaling pathway. Int. J. Cancer 2018, 142, 959–967.
- Gao, Z.W.; Wang, H.P.; Lin, F.; Wang, X.; Long, M.; Zhang, H.Z.; Dong, K. CD73 promotes proliferation and migration of human cervical cancer cells independent of its enzyme activity. BMC Cancer 2017, 17, 135.
- Lu, N.; Lin, T.; Wang, L.; Qi, M.; Liu, Z.; Dong, H.; Zhang, X.; Zhai, C.; Wang, Y.; Liu, L.; et al. Association of SOX4 regulated by tumor suppressor miR-30a with poor prognosis in low-grade chondrosarcoma. Tumour Biol. 2015, 36, 3843–3852.
- Cappellari, A.R.; Pillat, M.M.; Souza, H.D.; Dietrich, F.; Oliveira, F.H.; Figueiro, F.; Abujamra, A.L.; Roesler, R.; Lecka, J.; Sevigny, J.; et al. Ecto-5’-Nucleotidase overexpression reduces tumor growth in a xenograph medulloblastoma model. PLoS ONE 2015, 10, e0140996.
- Boyd-Tressler, A.M.; Lane, G.S.; Dubyak, G.R. Up-Regulated ectonucleotidases in fas-associated death domain protein- and receptor-interacting protein kinase 1-deficient jurkat leukemia cells counteract extracellular ATP/AMP accumulation via Pannexin-1 channels during chemotherapeutic drug-induced apoptosis. Mol. Pharm. 2017, 92, 30–47.
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857.
- Maj, T.; Wang, W.; Crespo, J.; Zhang, H.; Wang, W.; Wei, S.; Zhao, L.; Vatan, L.; Shao, I.; Szeliga, W.; et al. Oxidative stress controls regulatory T cell apoptosis and suppressor activity and PD-L1-blockade resistance in tumor. Nat. Immunol. 2017, 18, 1332–1341.
- Dumontet, C.; Peyrottes, S.; Rabeson, C.; Cros-Perrial, E.; Geant, P.Y.; Chaloin, L.; Jordheim, L.P. CD73 inhibition by purine cytotoxic nucleoside analogue-based diphosphonates. Eur. J. Med. Chem. 2018, 157, 1051–1055.
- Soleimani, A.; Bahreyni, A.; Roshan, M.K.; Soltani, A.; Ryzhikov, M.; Shafiee, M.; Soukhtanloo, M.; Jaafari, M.R.; Mashkani, B.; Hassanian, S.M. Therapeutic potency of pharmacological adenosine receptors agonist/antagonist on cancer cell apoptosis in tumor microenvironment, current status, and perspectives. J. Cell. Physiol. 2019, 234, 2329–2336.
- Tozzi, M.G.; Pesi, R.; Allegrini, S. On the physiological role of cytosolic 5’-nucleotidase II (cN-II): Pathological and therapeutical implications. Curr. Med. Chem. 2013, 20, 4285–4291.
- Galmarini, C.M.; Graham, K.; Thomas, X.; Calvo, F.; Rousselot, P.; El Jafaari, A.; Cros, E.; Mackey, J.R.; Dumontet, C. Expression of high Km 5’-nucleotidase in leukemic blasts is an independent prognostic factor in adults with acute myeloid leukemia. Blood 2001, 98, 1922–1926.
- Cividini, F.; Pesi, R.; Chaloin, L.; Allegrini, S.; Camici, M.; Cros-Perrial, E.; Dumontet, C.; Jordheim, L.P.; Tozzi, M.G. The purine analog fludarabine acts as a cytosolic 5’-nucleotidase II inhibitor. Biochem. Pharm. 2015, 94, 63–68.
- Jordheim, L.P.; Chaloin, L. Therapeutic perspectives for cN-II in cancer. Curr. Med. Chem. 2013, 20, 4292–4303.
- Jordheim, L.P.; Puy, J.Y.; Cros-Perrial, E.; Peyrottes, S.; Lefebvre, I.; Perigaud, C.; Dumontet, C. Determination of the enzymatic activity of cytosolic 5’-nucleotidase cN-II in cancer cells: Development of a simple analytical method and related cell line models. Anal. Bioanal. Chem. 2015, 407, 5747–5758.
- Cividini, F.; Cros-Perrial, E.; Pesi, R.; Machon, C.; Allegrini, S.; Camici, M.; Dumontet, C.; Jordheim, L.P.; Tozzi, M.G. Cell proliferation and drug sensitivity of human glioblastoma cells are altered by the stable modulation of cytosolic 5’-nucleotidase II. Int. J. Biochem. Cell Biol. 2015, 65, 222–229.
- Pesi, R.; Petrotto, E.; Colombaioni, L.; Allegrini, S.; Garcia-Gil, M.; Camici, M.; Jordheim, L.P.; Tozzi, M.G. Cytosolic 5’-Nucleotidase II silencing in a human lung carcinoma cell line opposes cancer phenotype with a concomitant increase in p53 phosphorylation. Int. J. Mol. Sci. 2018, 19, 2115.
- Bricard, G.; Cadassou, O.; Cassagnes, L.E.; Cros-Perrial, E.; Payen-Gay, L.; Puy, J.Y.; Lefebvre-Tournier, I.; Tozzi, M.G.; Dumontet, C.; Jordheim, L.P. The cytosolic 5’-nucleotidase cN-II lowers the adaptability to glucose deprivation in human breast cancer cells. Oncotarget 2017, 8, 67380–67393.
- Careddu, M.G.; Allegrini, S.; Pesi, R.; Camici, M.; Garcia-Gil, M.; Tozzi, M.G. Knockdown of cytosolic 5’-nucleotidase II (cN-II) reveals that its activity is essential for survival in astrocytoma cells. Biochim. Biophys. Acta 2008, 1783, 1529–1535.
- Kulkarni, S.S.; Karlsson, H.K.; Szekeres, F.; Chibalin, A.V.; Krook, A.; Zierath, J.R. Suppression of 5’-nucleotidase enzymes promotes AMP-activated protein kinase (AMPK) phosphorylation and metabolism in human and mouse skeletal muscle. J. Biol. Chem. 2011, 286, 34567–34574.
- Kviklyte, S.; Vertommen, D.; Yerna, X.; Andersen, H.; Xu, X.; Gailly, P.; Bohlooly, Y.M.; Oscarsson, J.; Rider, M.H. Effects of genetic deletion of soluble 5’-nucleotidases NT5C1A and NT5C2 on AMPK activation and nucleotide levels in contracting mouse skeletal muscles. Am. J. Physiol. Endocrinol. Metab. 2017, 313, e48–e62.
- Jordheim, L.P. Expanding the clinical relevance of the 5’-nucleotidase cN-II/NT5C2. Purinergic Signal. 2018, 14, 321–329.
- Cividini, F.; Filoni, D.N.; Pesi, R.; Allegrini, S.; Camici, M.; Tozzi, M.G. IMP-GMP specific cytosolic 5’-nucleotidase regulates nucleotide pool and prodrug metabolism. Biochim. Biophys. Acta 2015, 1850, 1354–1361.
- Allegrini, S.; Filoni, D.N.; Galli, A.; Collavoli, A.; Pesi, R.; Camici, M.; Tozzi, M.G. Expression of bovine cytosolic 5’-Nucleotidase (cN-II) in yeast: Nucleotide pools disturbance and its consequences on growth and homologous recombination. PLoS ONE 2013, 8, e63914.
- Gakis, C. Adenosine deaminase (ADA) isoenzymes ADA1 and ADA2: Diagnostic and biological role. Eur. Respir. J. 1996, 9, 632–633.
- Franco, R.; Casado, V.; Ciruela, F.; Saura, C.; Mallol, J.; Canela, E.I.; Lluis, C. Cell surface adenosine deaminase: Much more than an ectoenzyme. Prog. Neurobiol. 1997, 52, 283–294.
- Moreno, E.; Canet, J.; Gracia, E.; Lluis, C.; Mallol, J.; Canela, E.I.; Cortes, A.; Casado, V. Molecular evidence of adenosine deaminase linking adenosine A2A receptor and CD26 proteins. Front. Pharm. 2018, 9, 106.
- Biri, H.; Ozturk, S.; Kacmaz, M.; Karaca, K.; Tokucoglu, H.; Durak, I. Activities of DNA turnover and free radical metabolizing enzymes in cancerous human prostate tissue. Cancer Investig. 1999, 17, 314–319.
- Namiot, Z.; Stasiewicz, J.; Namiot, A.; Kemona, A.; Kralisz, M.; Gorski, J. Adenosine deaminase activity in patients with the intestinal type of gastric carcinoma. Cancer Lett. 1996, 109, 199–202.
- Specchia, G.; Pavone, V.; Maggio, F.; Lojudice, L.; Iacobazzi, A.; Detullio, L.; Cagnazzo, G.; Liso, V. Adenosine-Deaminase activity in peripheral lymphocytes of patients with gynecologic malignancies. Boll. Inst. Sieroter. Milan. 1985, 64, 404–407.
- Sufrin, G.; Tritsch, G.L.; Mittelman, A.; Moore, R.H.; Murphy, G.P. Adenosine-Deaminase activity in patients with renal adenocarcinoma. Cancer 1977, 40, 796–802.
- Dasmahapatra, K.S.; Hill, H.Z.; Dasmahapatra, A.; Suarez, S. Evaluation of adenosine-deaminase activity in patients with head and neck-cancer. J. Surg. Res. 1986, 40, 368–373.
- Kojima, O.; Majima, T.; Uehara, Y.; Yamane, T.; Fujita, Y.; Takahashi, T.; Majima, S. Alteration of adenosine-deaminase levels in peripheral-blood lymphocytes of patients with gastric-cancer. Jpn. J. Surg. 1985, 15, 130–133.
- Russo, M.; Giancane, R.; Apice, G.; Galanti, B. Adenosine-Deaminase and purine nucleoside phosphorylase activities in peripheral lymphocytes from patients with solid tumors. Br. J. Cancer 1981, 43, 196–200.
- Murray, J.L.; Perezsoler, R.; Bywaters, D.; Hersh, E.M. Decreased adenosine-deaminase (Ada) and 5’nucleotidase (5nt) activity in peripheral-blood T-Cells in Hodgkin disease. Am. J. Hematol. 1986, 21, 57–66.
- Camici, M.; Tozzi, M.G.; Allegrini, S.; Delcorso, A.; Sanfilippo, O.; Daidone, M.G.; Demarco, C.; Ipata, P.L. Purine salvage enzyme-activities in normal and neoplastic human tissues. Cancer Biochem. Bioph. 1990, 11, 201–209.
- Aghaei, M.; Karami-Tehrani, F.; Salami, S.; Atri, M. Adenosine deaminase activity in the serum and malignant tumors of breast cancer: The assessment of isoenzyme ADA1 and ADA2 activities. Clin. Biochem. 2005, 38, 887–891.
- Mahajan, M.; Tiwari, N.; Sharma, R.; Kaur, S.; Singh, N. Oxidative stress and its relationship with adenosine deaminase activity in various stages of breast cancer. Indian J. Clin. Biochem. 2013, 28, 51–54.
- Durak, I.; Beduk, Y.; Kavutcu, M.; Suzer, O.; Yaman, O.; Ozturk, H.S.; Canbolat, O.; Ulutepe, S. Activity of the enzymes participating in purine metabolism of cancerous and noncancerous human kidney tissues. Cancer Invest. 1997, 15, 212–216.
- Eroglu, A.; Canbolat, O.; Demirci, S.; Kocaoglu, H.; Eryavuz, Y.; Akgul, H. Activities of adenosine deaminase and 5 ‘-nucleotidase in cancerous and noncancerous human colorectal tissues. Med. Oncol. 2000, 17, 319–324.
- Pirincci, N.; Gecit, I.; Gunes, M.; Yuksel, M.B.; Kaba, M.; Tanik, S.; Demir, H.; Aslan, M. Serum adenosine deaminase, catalase and carbonic anhydrase activities in patients with bladder cancer. Clinics 2012, 67, 1443–1446.
- Urunsak, I.F.; Gulec, U.K.; Paydas, S.; Seydaoglu, G.; Guzel, A.B.; Vardar, M.A. Adenosine deaminase activity in patients with ovarian neoplasms. Arch. Gynecol. Obs. 2012, 286, 155–159.
- Sharma, S.D.; Desai, P.B.; Metgudmath, R.B. Evaluation of serum adenosine deaminase and retinol in patients with laryngeal cancer. Indian J. Pharm. Biol. Res. 2013, 1, 5.
- Lal, H.; Munjal, S.K.; Wig, U.; Saini, A.S. Serum enzymes in head and neck cancer III. J. Laryngol. Otol. 1987, 101, 1062–1065.
- Mishra, R.; Agarwal, M.K.; Chansuria, J.P. Serum adenosine deaminase levels as an index of tumor growth in head and neck malignancy. Indian J. Otolaryngol. Head Neck Surg. 2000, 52, 360–363.
- Ghaderi, B.; Amini, S.; Maroofi, F.; Jalali, C.; Javanmardi, M.; Roshani, D.; Abdi, M. Adenosine deaminase activity in chronic lymphocytic leukemia and healthy subjects. Iran. J. Cancer Prev. 2016, 9, e5069.
- Whitmore, K.V.; Gaspar, H.B. Adenosine deaminase deficiency—More than just an immunodeficiency. Front. Immunol. 2016, 7, 314.
- Agarwal, R.P. Recovery of 2’-deoxycoformycin-inhibited adenosine deaminase of mouse erythrocytes and leukemia L1210 in vivo. Cancer Res. 1979, 39, 1425–1427.
- Dohner, H.; Ho, A.D.; Thaler, J.; Stryckmans, P.; Sonneveld, P.; de Witte, T.; Lechner, K.; Lauria, F.; Bodewadt-Radzun, S.; Suciu, S.; et al. Pentostatin in prolymphocytic leukemia: Phase II trial of the European organization for research and treatment of cancer leukemia cooperative study group. J. Natl. Cancer Inst. 1993, 85, 658–662.
- Willis, C.R.; Goodrich, A.; Park, K.; Waselenko, J.K.; Lucas, M.; Reese, A.; Diehl, L.F.; Grever, M.R.; Byrd, J.C.; Flinn, I.W. A phase I/II study examining pentostatin, chlorambucil, and theophylline in patients with relapsed chronic lymphocytic leukemia and non-Hodgkin’s lymphoma. Ann. Hematol. 2006, 85, 301–307.
- Kay, N.E.; LaPlant, B.R.; Pettinger, A.M.; Call, T.G.; Leis, J.F.; Ding, W.; Parikh, S.A.; Conte, M.J.; Bowen, D.A.; Shanafelt, T.D. Cumulative experience and long term follow-up of pentostatin-based chemoimmunotherapy trials for patients with chronic lymphocytic leukemia. Expert Rev. Hematol. 2018, 11, 337–349.
- Tedeschi, A.; Rossi, D.; Motta, M.; Quaresmini, G.; Rossi, M.; Coscia, M.; Anastasia, A.; Rossini, F.; Cortelezzi, A.; Nador, G.; et al. A phase II multi-center trial of pentostatin plus cyclophosphamide with ofatumumab in older previously untreated chronic lymphocytic leukemia patients. Haematologica 2015, 100, e501–e504.
- Johnston, J.B. Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leuk. Lymphoma 2011, 52, 43–45.
- Hunt, S.W., 3rd; Hoffee, P.A. Adenosine deaminase from deoxycoformycin-sensitive and -resistant rat hepatoma cells. Purification and characterization. J. Biol. Chem. 1982, 257, 14239–14244.
- Camici, M.; Turriani, M.; Tozzi, M.G.; Turchi, G.; Cos, J.; Alemany, C.; Miralles, A.; Noe, V.; Ciudad, C.J. Purine enzyme profile in human colon-carcinoma cell-lines and differential sensitivity to deoxycoformycin and 2’-Deoxyadenosine in combination. Int. J. Cancer 1995, 62, 176–183.
- Bemi, V.; Tazzini, N.; Banditelli, S.; Giorgelli, F.; Pesi, R.; Turchi, C.; Mattana, A.; Sgarrella, F.; Tozzi, M.G.; Camici, M. Deoxyadenosine metabolism in a human colon-carcinoma cell line (LoVo) in relation to its cytotoxic effect in combination with deoxycoformycin. Int. J. Cancer 1998, 75, 713–720.
- Giannecchini, M.; D’Innocenzo, B.; Pesi, R.; Sgarrella, F.; Iorio, M.; Collecchi, P.; Tozzi, M.G.; Camici, M. 2 ‘-deoxyadenosine causes apoptotic cell death in a human colon carcinoma cell line. J. Biochem. Mol. Toxic 2003, 17, 329–337.
- Garcia-Gil, M.; Tozzi, M.G.; Allegrini, S.; Folcarelli, S.; Della Sala, G.; Voccoli, V.; Colombaioni, L.; Camici, M. Novel metabolic aspects related to adenosine deaminase inhibition in a human astrocytoma cell line. Neurochem. Int. 2012, 60, 523–532.
- Garcia-Gil, M.; Tozzi, M.G.; Balestri, F.; Colombaioni, L.; Camici, M. Mitochondrial damage and apoptosis induced by adenosine deaminase inhibition and deoxyadenosine in human neuroblastoma cell lines. J. Cell Biochem. 2016, 117, 1671–1679.
- Garcia-Gil, M.; Tozzi, M.G.; Varani, S.; Della Verde, L.; Petrotto, E.; Balestri, F.; Colombaioni, L.; Camici, M. The combination of adenosine deaminase inhibition and deoxyadenosine induces apoptosis in a human astrocytoma cell line. Neurochem. Int. 2015, 80, 14–22.
- Thirupathi, A.; Chang, Y.Z. Role of AMPK and its molecular intermediates in subjugating cancer survival mechanism. Life Sci. 2019, 227, 30–38.
- Saito, M.; Yaguchi, T.; Yasuda, Y.; Nakano, T.; Nishizaki, T. Adenosine suppresses CW2 human colonic cancer growth by inducing apoptosis via A(1) adenosine receptors. Cancer Lett. 2010, 290, 211–215.
- Sai, K.; Yang, D.; Yamamoto, H.; Fujikawa, H.; Yamamoto, S.; Nagata, T.; Saito, M.; Yamamura, T.; Nishizaki, T. A(1) adenosine receptor signal and AMPK involving caspase-9/-3 activation are responsible for adenosine-induced RCR-1 astrocytoma cell death. Neurotoxicology 2006, 27, 458–467.
- Chen, Y.; Yang, S.H.; Hueng, D.Y.; Syu, J.P.; Liao, C.C.; Wu, Y.C. Cordycepin induces apoptosis of C6 glioma cells through the adenosine 2A receptor-p53-caspase-7-PARP pathway. Chem. Biol. Interact. 2014, 216, 17–25.
- Merighi, S.; Mirandola, P.; Milani, D.; Varani, K.; Gessi, S.; Klotz, K.N.; Leung, E.; Baraldi, P.G.; Borea, P.A. Adenosine receptors as mediators of both cell proliferation and cell death of cultured human melanoma cells. J. Invest. Derm. 2002, 119, 923–933.
- Tamura, K.; Kanno, T.; Fujita, Y.; Gotoh, A.; Nakano, T.; Nishizaki, T. A(2a) adenosine receptor mediates HepG2 cell apoptosis by downregulating Bcl-X(L) expression and upregulating Bid expression. J. Cell Biochem. 2012, 113, 1766–1775.
- Yasuda, Y.; Saito, M.; Yamamura, T.; Yaguchi, T.; Nishizaki, T. Extracellular adenosine induces apoptosis in Caco-2 human colonic cancer cells by activating caspase-9/-3 via A(2a) adenosine receptors. J. Gastroenterol. 2009, 44, 56–65.
- Hajiahmadi, S.; Panjehpour, M.; Aghaei, M.; Shabani, M. Activation of A2b adenosine receptor regulates ovarian cancer cell growth: Involvement of Bax/Bcl-2 and caspase-3. Biochem. Cell Biol. 2015, 93, 321–329.
- Jafari, S.M.; Joshaghani, H.R.; Panjehpour, M.; Aghaei, M. A2B adenosine receptor agonist induces cell cycle arrest and apoptosis in breast cancer stem cells via ERK1/2 phosphorylation. Cell Oncol. 2018, 41, 61–72.
- Abedi, H.; Aghaei, M.; Panjehpour, M.; Hajiahmadi, S. Mitochondrial and caspase pathways are involved in the induction of apoptosis by IB-MECA in ovarian cancer cell lines. Tumour Biol. 2014, 35, 11027–11039.
- Jafari, S.M.; Panjehpour, M.; Aghaei, M.; Joshaghani, H.R.; Enderami, S.E. A3 adenosine receptor agonist inhibited survival of breast cancer stem cells via GLI-1 and ERK1/2 pathway. J. Cell Biochem. 2017, 118, 2909–2920.
- Cohen, S.; Stemmer, S.M.; Zozulya, G.; Ochaion, A.; Patoka, R.; Barer, F.; Bar-Yehuda, S.; Rath-Wolfson, L.; Jacobson, K.A.; Fishman, P. CF102 an A3 adenosine receptor agonist mediates anti-tumor and anti-inflammatory effects in the liver. J. Cell. Physiol. 2011, 226, 2438–2447.
- Kanno, T.; Gotoh, A.; Fujita, Y.; Nakano, T.; Nishizaki, T. A(3) adenosine receptor mediates apoptosis in 5637 human bladder cancer cells by G(q) protein/PKC-dependent AIF upregulation. Cell. Physiol. Biochem. 2012, 30, 1159–1168.
- Kanno, T.; Nakano, T.; Fujita, Y.; Gotoh, A.; Nishizaki, T. Adenosine induces apoptosis in SBC-3 human lung cancer cells through A(3) adenosine receptor-dependent AMID upregulation. Cell. Physiol. Biochem. 2012, 30, 666–677.
- Nagaya, H.; Gotoh, A.; Kanno, T.; Nishizaki, T. A3 adenosine receptor mediates apoptosis in in vitro RCC4-VHL human renal cancer cells by up-regulating AMID expression. J. Urol. 2013, 189, 321–328.
- Aghaei, M.; Panjehpour, M.; Karami-Tehrani, F.; Salami, S. Molecular mechanisms of A3 adenosine receptor-induced G1 cell cycle arrest and apoptosis in androgen-dependent and independent prostate cancer cell lines: Involvement of intrinsic pathway. J. Cancer Res. Clin. Oncol. 2011, 137, 1511–1523.
- Otsuki, T.; Kanno, T.; Fujita, Y.; Tabata, C.; Fukuoka, K.; Nakano, T.; Gotoh, A.; Nishizaki, T. A3 adenosine receptor-mediated p53-dependent apoptosis in Lu-65 human lung cancer cells. Cell. Physiol. Biochem. 2012, 30, 210–220.
- Kim, S.G.; Ravi, G.; Hoffmann, C.; Jung, Y.J.; Kim, M.; Chen, A.; Jacobson, K.A. p53-Independent induction of Fas and apoptosis in leukemic cells by an adenosine derivative, Cl-IB-MECA. Biochem. Pharm. 2002, 63, 871–880.
- Jiang, X.; Tan, H.Y.; Teng, S.; Chan, Y.T.; Wang, D.; Wang, N. The role of AMP-Activated protein kinase as a potential target of treatment of hepatocellular carcinoma. Cancers 2019, 11, 647.
- Tsuchiya, A.; Nishizaki, T. Anticancer effect of adenosine on gastric cancer via diverse signaling pathways. World J. Gastroenterol. 2015, 21, 10931–10935.
- Saitoh, M.; Nagai, K.; Nakagawa, K.; Yamamura, T.; Yamamoto, S.; Nishizaki, T. Adenosine induces apoptosis in the human gastric cancer cells via an intrinsic pathway relevant to activation of AMP-Activated protein kinase. Biochem. Pharmacol. 2004, 67, 2005–2011.
- Nogi, Y.; Kanno, T.; Nakano, T.; Fujita, Y.; Tabata, C.; Fukuoka, K.; Gotoh, A.; Nishizaki, T. AMP converted from intracellularly transported adenosine upregulates p53 expression to induce malignant pleural mesothelioma cell apoptosis. Cell. Physiol. Biochem. 2012, 30, 61–74.
- Zuckerman, V.; Wolyniec, K.; Sionov, R.V.; Haupt, S.; Haupt, Y. Tumour suppression by p53: The importance of apoptosis and cellular senescence. J. Pathol. 2009, 219, 3–15.
- Nakajima, Y.; Kanno, T.; Nagaya, T.; Kuribayashi, K.; Nakano, T.; Gotoh, A.; Nishizaki, T. Adenosine deaminase inhibitor EHNA exhibits a potent anticancer effect against malignant pleural mesothelioma. Cell. Physiol. Biochem. 2015, 35, 51–60.
- Haynes, J.; Killilea, D.W.; Peterson, P.D.; Thompson, W.J. Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic-3’,5’-guanosine monophosphate-stimulated phosphodiesterase to reverse hypoxic pulmonary vasoconstriction in the perfused rat lung. J. Pharm. Exp. 1996, 276, 752–757.
- Tsuchiya, A.; Kanno, T.; Saito, M.; Miyoshi, Y.; Gotoh, A.; Nakano, T.; Nishizaki, T. Intracellularly transported adenosine induces apoptosis in MCF-7 human breast cancer cells by accumulating AMID in the nucleus. Cancer Lett. 2012, 321, 65–72.
- Bano, D.; Prehn, J.H.M. Apoptosis-Inducing factor (AIF) in physiology and disease: The tale of a repented natural born killer. EBioMedicine 2018, 30, 29–37.
- Yang, D.; Yaguchi, T.; Nagata, T.; Gotoh, A.; Dovat, S.; Song, C.; Nishizaki, T. AMID mediates adenosine-induced caspase-independent HuH-7 cell apoptosis. Cell Physiol. Biochem. 2011, 27, 37–44.
- Hermes, M.; Osswald, H.; Kloor, D. Role of S-adenosylhomocysteine hydrolase in adenosine-induced apoptosis in HepG2 cells. Exp. Cell Res. 2007, 313, 264–283.
- Kutryb-Zajac, B.; Koszalka, P.; Mierzejewska, P.; Bulinska, A.; Zabielska, M.A.; Brodzik, K.; Skrzypkowska, A.; Zelazek, L.; Pelikant-Malecka, I.; Slominska, E.M.; et al. Adenosine deaminase inhibition suppresses progression of 4T1 murine breast cancer by adenosine receptor-dependent mechanisms. J. Cell Mol. Med. 2018, 22, 5939–5954.


