 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Junnian Wei | + 2086 word(s) | 2086 | 2021-09-29 02:59:37 | | | |
| 2 | Conner Chen | Meta information modification | 2086 | 2021-10-08 10:52:31 | | |
Video Upload Options
Copper-catalyzed and organocopper-involved reactions are of great significance in organic synthesis. To have a deep understanding of the reaction mechanisms, the structural characterizations of organocopper intermediates become indispensable. Meanwhile, the structure-function relationship of organocopper compounds could advance the rational design and development of new Cu-based reactions and organocopper reagents. Compared to the mono-carbonic ligand, the C,N- and C,C-bidentate ligands better stabilize unstable organocopper compounds. Bidentate ligands can chelate to the same copper atom via η2-mode, forming a mono-cupra-cyclic compounds with at least one acute C-Cu-C angle. When the bidentate ligands bind to two copper atoms via η1-mode at each coordinating site, the bimetallic macrocyclic compounds will form nearly linear C-Cu-C angles. The anionic coordinating sites of the bidentate ligand can also bridge two metals via μ2-mode, forming organocopper aggregates with Cu-Cu interactions and organocuprates with contact ion pair structures. The reaction chemistry of some selected organocopper compounds is highlighted, showing their unique structure–reactivity relationships.
1. Introduction
- (1)
-
Mononuclear organocopper complexes:
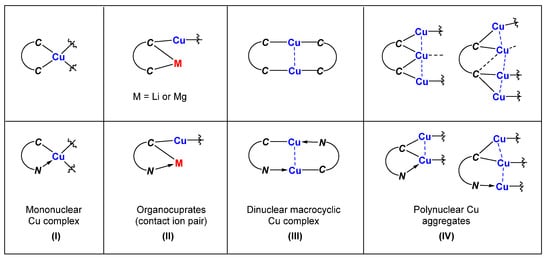
- (2)
-
Organocuprates with contact ion pair structures:
- (3)
-
Binuclear macrocyclic copper complexes:
- (4)
-
Polynuclear organocopper aggregates:
2. Mononuclear Organocopper Complexes
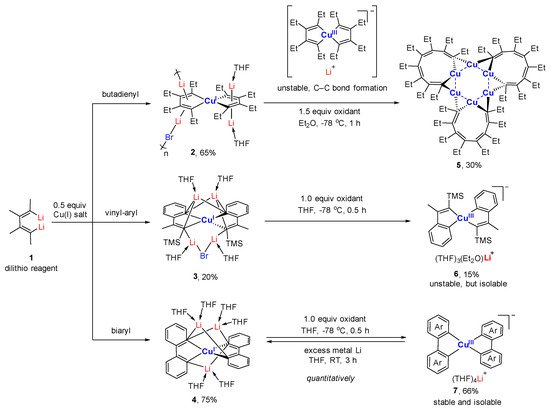
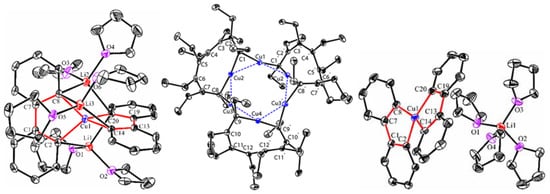
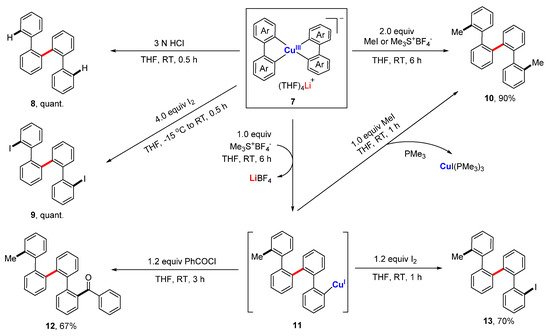
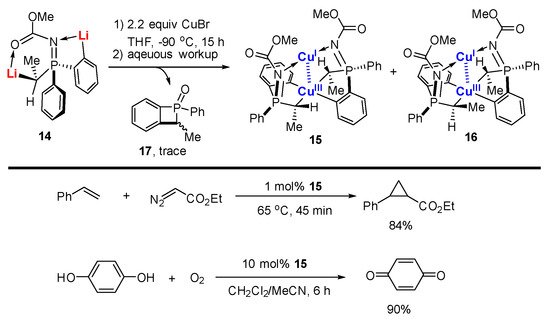
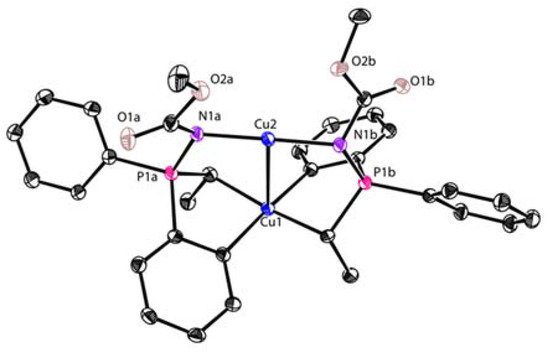
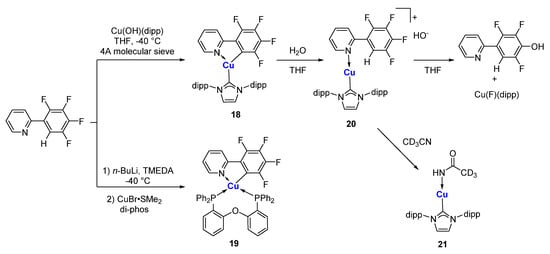
References
- Ley, S.V.; Thomas, A.W. Modern Synthetic Methods for Copper-Mediated C(aryl)–O, C(aryl)–N, and C(aryl)–S Bond Formation. Angew. Chem. Int. Ed. 2003, 42, 5400–5449.
- Evano, G.; Blanchard, N.; Toumi, M. Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis. Chem. Rev. 2008, 108, 3054–3131.
- Meldal, M.; Tornøe, C.W. Cu-Catalyzed Azide–Alkyne Cycloaddition. Chem. Rev. 2008, 108, 2952–3015.
- Sperotto, E.; van Klink, G.P.M.; van Koten, G.; de Vries, J.G. The mechanism of the modified Ullmann reaction. Dalton Trans. 2010, 39, 10338–10351.
- Beletskaya, I.P.; Cheprakov, A.V. The Complementary Competitors: Palladium and Copper in C–N Cross-Coupling Reactions. Organometallics 2012, 31, 7753–7808.
- Gephart, R.T.; Warren, T.H. Copper-Catalyzed sp3 C–H Amination. Organometallics 2012, 31, 7728–7752.
- Zhang, C.; Tang, C.; Jiao, N. Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process. Chem. Soc. Rev. 2012, 41, 3464–3484.
- Allen, S.E.; Walvoord, R.R.; Padilla-Salinas, R.; Kozlowski, M.C. Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev. 2013, 113, 6234–6458.
- Evano, G.; Blanchard, N. Copper-Mediated Cross-Coupling Reactions; Evano, G., Blanchard, N., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2013.
- Lazreg, F.; Nahra, F.; Cazin, C.S.J. Copper–NHC complexes in catalysis. Coord. Chem. Rev. 2015, 293–294, 48–79.
- McCann, S.D.; Stahl, S.S. Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox-Active Catalyst. Acc. Chem. Res. 2015, 48, 1756–1766.
- Jordan, A.J.; Lalic, G.; Sadighi, J.P. Coinage Metal Hydrides: Synthesis, Characterization, and Reactivity. Chem. Rev. 2016, 116, 8318–8372.
- Lipshutz, B.H.; Sengupta, S. Organocopper Reagents: Substitution, Conjugate Addition, Carbo/Metallocupration, and Other Reactions. Org. React. 1992, 135–631.
- Surry, D.S.; Spring, D.R. The oxidation of organocuprates-an offbeat strategy for synthesis. Chem. Soc. Rev. 2006, 35, 218–225.
- Breit, B.; Schmidt, Y. Directed Reactions of Organocopper Reagents. Chem. Rev. 2008, 108, 2928–2951.
- Ullenius, C.; Christenson, B.L. Organocuprate addition to α,β-unsaturated compounds: Synthetic and mechanistic aspects. Pure Appl. Chem. 1988, 60, 57–64.
- Woodward, S. Decoding the ‘black box’ reactivity that is organocuprate conjugate addition chemistry. Chem. Soc. Rev. 2000, 29, 393–401.
- Kharasch, M.S.; Tawney, P.O. Factors Determining the Course and Mechanisms of Grignard Reactions. II. The Effect of Metallic Compounds on the Reaction between Isophorone and Methylmagnesium Bromide. J. Am. Chem. Soc. 1941, 63, 2308–2316.
- Krause, N. Modern Organocopper Chemistry; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2002.
- Rappoport, Z.; Marek, I. The Chemistry of Organocopper Compounds; John Wiley & Sons Ltd.: Chichester, UK, 2009.
- Piazza, C.; Knochel, P. Sterically Hindered Lithium Dialkylcuprates for the Generation of Highly Functionalized Mixed Cuprates through a Halogen–Copper Exchange. Angew. Chem. Int. Ed. 2002, 41, 3263–3265.
- Zanardi, A.; Novikov, M.A.; Martin, E.; Benet-Buchholz, J.; Grushin, V.V. Direct Cupration of Fluoroform. J. Am. Chem. Soc. 2011, 133, 20901–20913.
- Ebert, G.; Rieke, R.D. Direct formation of organocopper compounds by oxidative addition of zerovalent copper to organic halides. J. Org. Chem. 1984, 49, 5280–5282.
- Gilman, H.; Jones, R.G.; Woods, L.A. The Preparation of Methylcopper and some Observations on the Decomposition of Organocopper Compounds. J. Org. Chem. 1952, 17, 1630–1634.
- Jukes, A.E. The Organic Chemistry of Copper. In Advances in Organometallic Chemistry; Stone, F.G.A., West, R., Eds.; Academic Press: New York, NY, USA, 1974; Volume 12, pp. 215–322.
- Costa, G.; Camus, A.; Gatti, L.; Marsich, N. On phenylcopper. J. Organomet. Chem. 1966, 5, 568–572.
- Stephens, R.D.; Castro, C.E. The Substitution of Aryl Iodides with Cuprous Acetylides. A Synthesis of Tolanes and Heterocyclics. J. Org. Chem. 1963, 28, 3313–3315.
- van Koten, G. A view of organocopper compound and cuprates. J. Organomet. Chem. 1990, 400, 283–301.
- van Koten, G. Organocopper Compounds: From Elusive to Isolable Species, from Early Supramolecular Chemistry with RCuI Building Blocks to Mononuclear R2–nCuII and R3–mCuIII Compounds. A Personal View. Organometallics 2012, 31, 7634–7646.
- Zhang, S.; Zhang, W.-X.; Xi, Z. Organo-di-Lithio Reagents: Cooperative Effect and Synthetic Applications. In Organo-di-Metallic Compounds (or Reagents): Synergistic Effects and Synthetic Applications; Xi, Z., Ed.; Springer International Publishing: Cham, Switzerland, 2014; pp. 1–41.
- Zhang, W.-X.; Xi, Z. Organometallic intermediate-based organic synthesis: Organo-di-lithio reagents and beyond. Org. Chem. Front. 2014, 1, 1132–1139.
- Wei, J.; Zhang, Y.; Chi, Y.; Liu, L.; Zhang, W.-X.; Xi, Z. Aromatic Dicupraannulenes. J. Am. Chem. Soc. 2016, 138, 60–63.
- Green, J.C.; Green, M.L.H.; Parkin, G. The occurrence and representation of three-centre two-electron bonds in covalent inorganic compounds. Chem. Commun. 2012, 48, 11481–11503.
- Zhou, Y.; Zhang, W.-X.; Xi, Z. 1,3-Butadienylzinc Trimer Formed via Transmetalation from 1,4-Dilithio-1,3-butadienes: Synthesis, Structural Characterization, and Application in Negishi Cross-Coupling. Organometallics 2012, 31, 5546–5550.
- Wei, J.; Liu, L.; Zhan, M.; Xu, L.; Zhang, W.-X.; Xi, Z. Magnesiacyclopentadienes as Alkaline-Earth Metallacyclopentadienes: Facile Synthesis, Structural Characterization, and Synthetic Application. Angew. Chem. Int. Ed. 2014, 53, 5634–5638.
- Wei, J.; Zhang, W.-X.; Xi, Z. Dianions as Formal Oxidants: Synthesis and Characterization of Aromatic Dilithionickeloles from 1,4-Dilithio-1,3-butadienes and . Angew. Chem. Int. Ed. 2015, 54, 5999–6002.
- Wei, J.; Zhang, Y.; Zhang, W.-X.; Xi, Z. 1,3-Butadienyl Dianions as Non-Innocent Ligands: Synthesis and Characterization of Aromatic Dilithio Rhodacycles. Angew. Chem. Int. Ed. 2015, 54, 9986–9990.
- Xu, L.; Wang, Y.C.; Wei, J.; Wang, Y.; Wang, Z.; Zhang, W.-X.; Xi, Z. The First Lutetacyclopentadienes: Synthesis, Structure, and Diversified Insertion/C–H Activation Reactivity. Chem. Eur. J. 2015, 21, 6686–6689.
- Zhang, Y.; Wei, J.; Zhang, W.-X.; Xi, Z. Lithium Aluminate Complexes and Alumoles from 1,4-Dilithio-1,3-Butadienes and AlEt2Cl. lnorg. Chem. 2015, 54, 10695–10700.
- Xu, L.; Wang, Y.; Wang, Y.-C.; Wang, Z.; Zhang, W.-X.; Xi, Z. Sandwich Lutetacyclopentadiene with the Coordination of Lithium to the Diene Unit: Synthesis, Structure, and Transformation. Organometallics 2016, 35, 5–8.
- Ma, W.; Yu, C.; Chi, Y.; Chen, T.; Wang, L.; Yin, J.; Wei, B.; Xu, L.; Zhang, W.-X.; Xi, Z. Formation and ligand-based reductive chemistry of bridged bis-alkylidene scandium(III) complexes. Chem. Sci. 2017, 8, 6852–6856.
- Wei, B.; Liu, L.; Zhang, W.-X.; Xi, Z. Synthesis and Structural Characterization of Butadienylcalcium-based Heavy Grignard Reagents and a Ca4 Inverse Crown Ether Complex. Angew. Chem. Int. Ed. 2017, 56, 9188–9192.
- Zhang, Y.; Chi, Y.; Wei, J.; Yang, Q.; Yang, Z.; Chen, H.; Yang, R.; Zhang, W.-X.; Xi, Z. Aromatic Tetralithiodigalloles with a Ga–Ga Bond: Synthesis and Structural Characterization. Organometallics 2017, 36, 2982–2986.
- Zhang, Y.; Wei, J.; Chi, Y.; Zhang, X.; Zhang, W.-X.; Xi, Z. Spiro Metalla-aromatics of Pd, Pt, and Rh: Synthesis and Characterization. J. Am. Chem. Soc. 2017, 139, 5039–5042.
- Zhu, M.; Liu, L.; Zhang, Y.; Yu, H.T.; Zhang, W.-X.; Xi, Z. Selective Transformation of Well-Defined Alkenyllithiums to Alkenylmagnesiums via Transmetalation. Chem. Eur. J. 2017, 24, 3186–3191.
- Wei, B.; Zhang, W.-X.; Xi, Z. Well-defined styryl and biphenyl calcium complexes from dilithio compounds and calcium iodide: Synthesis, structure and reactivity toward nitrous oxide. Dalton Trans. 2018, 47, 12540–12545.
- Huang, Z.; Zhang, Y.; Zhang, W.-X.; Xi, Z. Reversible Two-Electron Redox Reactions Involving Tetralithio/Dilithio Palladole, Platinacycle, and Dicupraannulene. Organometallics 2019, 38, 2807–2811.
- Zhang, Y.; Liu, L.; Chen, T.; Huang, Z.; Zhang, W.-X.; Xi, Z. Dilithio Spiro Zincacyclopentadienes and Dizincacycles: Synthesis and Structural Characterization. Organometallics 2019, 38, 2174–2178.
- Zhang, Y.; Wei, J.; Zhu, M.; Chi, Y.; Zhang, W.-X.; Ye, S.; Xi, Z. Tetralithio Metalla-aromatics with Two Independent Perpendicular Dilithio Aromatic Rings Spiro-fused by One Manganese Atom. Angew. Chem. Int. Ed. 2019, 58, 9625–9631.
- Zhang, Y.; Yang, Z.; Zhang, W.-X.; Xi, Z. Indacyclopentadienes and Aromatic Indacyclopentadienyl Dianions: Synthesis and Characterization. Chem. Eur. J. 2019, 25, 4218–4224.
- Yu, C.; Ma, W.; Zhang, W.-X.; Xi, Z. Mono- and Bis-Titanium Complexes Bridged by 2-Butene Tetraanion: Synthesis and Structural Characterization. Organometallics 2020, 39, 793–796.
- Yu, C.; Zhong, M.; Zhang, Y.; Wei, J.; Ma, W.; Zhang, W.-X.; Ye, S.; Xi, Z. Butadienyl Diiron Complexes: Nonplanar Metalla-Aromatics Involving σ-Type Orbital Overlap. Angew. Chem. Int. Ed. 2020, 59, 19048–19053.
- Zhang, Y.; Wu, B.; Zhong, M.; Zhang, W.-X.; Xi, Z. Cyclic Bis-alkylidene Complexes of Titanium and Zirconium: Synthesis, Characterization, and Reaction. Chem. Eur. J. 2020, 26, 16472–16479.
- Zheng, Y.; Cao, C.-S.; Ma, W.; Chen, T.; Wu, B.; Yu, C.; Huang, Z.; Yin, J.; Hu, H.-S.; Li, J.; et al. 2-Butene Tetraanion Bridged Dinuclear Samarium(III) Complexes via Sm(II)-Mediated Reduction of Electron-Rich Olefins. J. Am. Chem. Soc. 2020, 142, 10705–10714.
- Huang, Z.; Zhang, Y.; Zhang, W.-X.; Wei, J.; Ye, S.; Xi, Z. A tris-spiro metalla-aromatic system featuring Craig-Möbius aromaticity. Nat. Commun. 2021, 12, 1319.
- Geng, W.; Wei, J.; Zhang, W.-X.; Xi, Z. Isolable and Well-Defined Butadienyl Organocopper(I) Aggregates: Facile Synthesis, Structural Characterization, and Reaction Chemistry. J. Am. Chem. Soc. 2014, 136, 610–613.
- Liu, L.; Geng, W.; Yang, Q.; Zhang, W.-X.; Xi, Z. Well-Defined Butadienyl Organocopper(I) Aggregates from Zirconacyclopentadienes and CuCl: Synthesis and Structural Characterization. Organometallics 2015, 34, 4198–4201.
- Liu, L.; Wei, J.; Chi, Y.; Zhang, W.-X.; Xi, Z. Structure and Reaction Chemistry of Magnesium Organocuprates Derived from Magnesiacyclopentadienes and Copper(I) Salts. Angew. Chem. Int. Ed. 2016, 55, 14762–14765.
- Liu, L.; Zhu, M.; Yu, H.-T.; Zhang, W.-X.; Xi, Z. Organocopper(III) Spiro Complexes: Synthesis, Structural Characterization, and Redox Transformation. J. Am. Chem. Soc. 2017, 139, 13688–13691.
- Liu, L.; Zhu, M.; Yu, H.-T.; Zhang, W.-X.; Xi, Z. Formation of a Hexanuclear Octatetraenyl Organocopper(I) Aggregate via Oxidation of Spiro Butadienyl Organocuprate. Organometallics 2018, 37, 845–847.
- DiMucci, I.M.; Lukens, J.T.; Chatterjee, S.; Carsch, K.M.; Titus, C.J.; Lee, S.J.; Nordlund, D.; Betley, T.A.; MacMillan, S.N.; Lancaster, K.M. The Myth of d8 Copper(III). J. Am. Chem. Soc. 2019, 141, 18508–18520.
- García-López, J.; Yañez-Rodríguez, V.; Roces, L.; García-Granda, S.; Martínez, A.; Guevara-García, A.; Castro, G.R.; Jiménez-Villacorta, F.; Iglesias, M.J.; López-Ortiz, F. Synthesis and Characterization of a Coupled Binuclear CuI/CuIII Complex. J. Am. Chem. Soc. 2010, 132, 10665–10667.
- Molteni, R.; Edkins, K.; Haehnel, M.; Steffen, A. C–H Activation of Fluoroarenes: Synthesis, Structure, and Luminescence Properties of Copper(I) and Gold(I) Complexes Bearing 2-Phenylpyridine Ligands. Organometallics 2016, 35, 629–640.


