1. Introduction
Copper-mediated or -catalyzed reactions have been upgrading the toolbox of organic synthesis with a variety of cheap, efficient transformations, which benefit scientific research in a broad range of areas, such as in polymer chemistry and biochemistry
[1][2][3][4][5][6][7][8][9][10][11][12][1,2,3,4,5,6,7,8,9,10,11,12]. To have a deep understanding of the reaction mechanisms, organocopper species, as proposed intermediates, are sought to be isolated. On the other hand, organocopper compounds can be easily prepared in situ and utilized as organometallic synthons to construct various small organic molecules
[13][14][15][13,14,15]. A well-recognized example is that “hard” nucleophiles, e.g., organolithium or Grignard reagents, react with α,β-unsaturated carbonyl compounds via 1,2-addition, whereas the “soft” organocuprates will end up with 1,4-addition products with the same substrates
[16][17][16,17]. In a catalytic version, in the presence of copper salts, Grignard reagents can behave similarly to organocuprates
[18]. It is known that reactivities are closely connected to structural configurations. Organocopper chemistry focuses on establishing the structure–reactivity relationship of well-defined organocopper compounds, rationalizing the dissimilar reaction patterns of different organometallic copper reagents.
Transmetalation provides the most straightforward and reliable method f preparing organocopper compounds from readily available organometallic reagents
[19][20][19,20], though many other synthetic strategies are also known, such as copper-halide exchange reactions
[21], direct cupration
[22], and the oxidative addition of reactive Rieke Cu*
[23]. It should be emphasized that the identity of forming organocopper compounds is very much dependent on the ratio of starting materials. For example, in the early 19th century, Gilman et al. found that the reaction of methyl lithium and 1.0 equiv. of copper(I) salts formed insoluble methylcopper polymers as yellow solids in diethyl ether
[24]. When 0.5 equiv. of copper(I) salts was used in the above condition, a clear solution of dimethyl cuprate (Gilman reagent) was created. The initial isolation and characterizations of organocopper compounds were very slow since the reactive species were subjected to thermally homolytic, oxidative, and hydrolytic decompositions
[25]. A general trend is that the stability increases from alkyl, alkenyl, and aryl to alkynyl copper compounds. For example, methylcopper
[24] and phenylcopper
[26] clusters display rapid and slow decompositions at room temperature, respectively, while (phenylethynyl)copper
[27] looks stable under ambient conditions. This stability trend could also be reflected by the numbers of single-crystal structures in the Cambridge Structural Database (CSD 5.42, 2020 November), in which roughly 185 alkyl, 288 aryl, and 326 alkynyl copper compounds have been collected. Moreover, the aggregate state, which is favorable in charge-neutral organocopper compounds, makes these polymeric compounds poorly soluble in common organic solvents. To solve the above inherent problems of organocopper compounds, coordinating heteroatom-containing moieties, such as dimethyl amino groups, were rationally introduced into the ligand skeleton by van Koten and other researches to enhance stability and solubility
[28][29][28,29]. The hybrid C,N-bidentate ligands can bind to copper atoms with an enhanced chelating effect. Following a different path, Xi’s group discovered that 1,4-dilithio 1,3-butadienes (C,C-bidentate ligand) can stabilize reactive organocopper species to a great extent via a cooperative effect
[30][31][30,31]. In addition, this C,C-bidentate ligand can also be regarded as an analogue of 1,2-diketimine, which turns out to be a special Z-type non-innocent ligand, producing an unprecedented aromatic dicupro-annulenes
[32].
Here, in this review, we are interested in summarizing organocopper compounds with at least one Cu-C
σ-bond in which the organic fragments behave as actor ligands rather than spectator ligands (e.g., Cp, NHC). For mono-dentate carbonic ligands, the binding between copper and carbon atoms normally consists of the regular two-center two-electron (2c-2e)
σ-bonds and three-center two-electron (3c-2e) bonds
[33] when the carbon atom bridges two copper atoms. In cases where the bridging carbon atom is linked with a copper and a lithium (or magnesium) atom, the binding is more likely between the 2c-2e and 3c-2e bonds. For neutral nitrogen-containing groups, the binding of Cu-N is a traditional dative bond. When extending from one coordinating site to C,N- or C,C-chelating ligands, the binding in the corresponding organocopper compounds becomes more diverse. Based on the
characteristic connectivity of the fragments in these structures, the C,N- and C,C-chelated organocopper compounds can be put into the following four categories:
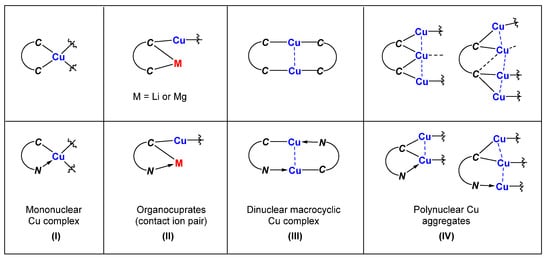
- (1)
-
Mononuclear organocopper complexes:
The two coordinating sites of the C,C- and C,N-bidentate ligand bind to the same Cu center together, forming a 5-membered chelate ring, as shown in
(I) of
Scheme 1. The two chelate rings are connected with a copper atom and will generate a spiro complex.
Scheme 1. Fragments in the molecular structures with characteristic connectivity. The lines do not necessarily represent 2c-2e or 3c-2e bonds.
- (2)
-
Organocuprates with contact ion pair structures:
The two coordinating sites can be attached with different metals, with one site bound to the Cu/M atoms and the other one bound to the main group metal atoms M only. The resulting product can be regarded as being organocuprates with contact ion pair structures (II).
- (3)
-
Binuclear macrocyclic copper complexes:
Each coordinating site of the C,C- and C,N-bidentate ligand can also bind an individual Cu atom with 2c-2e bonds, forming a macrocyclic dinuclear complex. As a result, each Cu atom forms a linear geometry with two units of bidentate ligands that provide only one coordinating site to each Cu atom (III).
- (4)
-
Polynuclear organocopper aggregates:
If the carbanion bridges two copper atoms, the structural feature will likely be polynuclear copper aggregates with Cu-Cu interactions (IV).
2. Mononuclear Organocopper Complexes
Although neutral N,N-chelated spiro copper complexes are relatively common, the anionic C,N- or C,C-cheated spiro counterparts are sporadic. As mentioned above, Xi’s group found that the C,C-bidentate ligand is an excellent platform to build up diversified coordination complexes across the periodic table
[32][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][52][53][54][55][56][57][58][59][60][32,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60]. The chelating effect increases the thermal stability of these organometallic species, facilitating isolation and following characterization. Additionally, their metal–carbon bonds remain reactive toward electrophiles. As a result, a good balance between stability and reactivity is achieved.
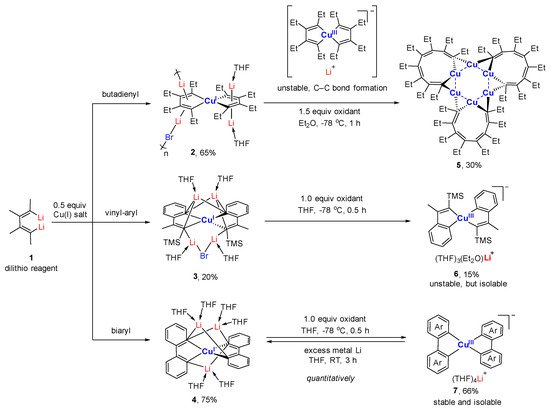
Starting from the 1,4-dilithio 1,3-butadienes
1 (dilithio reagent for short) and 0.5 equiv. of copper(I) salts, Xi’s group isolated the first series of spiro organocopper(I) compounds
2–4 (
Scheme 2)
[59][60][59,60]. It seems that the terminal alkenyl carbon atom is favorable for LiBr coordination, probably because it is more electron-rich than the phenyl ones. The butadienyl spiro copper complex
2 forms one-dimensional polymers with repeating units linked by Li—Br. When one vinyl moiety is replaced by a phenyl ring, LiBr forms a Li
2Br fragment between the two vinyl carbon atoms of
3. No LiBr salt can be found in the biphenyl spiro copper complex
4 (
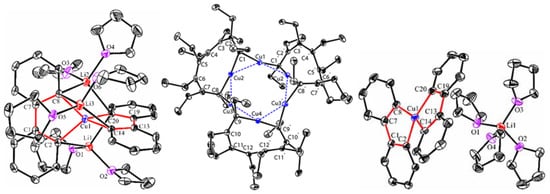
Figure 1). The copper atoms are all coordinated with two dianionic ligands, forming a distorted tetrahedral geometry (dihedral angels between two five-membered chelate rings is about 63–84°). The averaged Cu(I)-C bond lengths of
2–4 are from 2.01 to 2.06 Å. Upon oxidation, either the dianionic ligand or the metal center can be oxidized depending on the electron-richness of the ligands. Compound
1 is transformed to a hexanuclear tetraenyl copper cluster
5 with a Cu
6 hexagon. In this process, the oxidative dimerization of the original butadienyl ligand occurs. It might undergo a spiro Cu(III) intermediate, which is too unstable to be isolated. When
3 is treated with oxidizing agents, a spiro Cu(III) complex
6 is afforded and isolated successfully. Compound
6 is thermally unstable and is difficult to handle at room temperature. With an extended conjugated backbone,
4 is oxidized to form a very stable spiro Cu(III) complex
7. In addition,
7 can be reduced by metal lithium to regenerate
4 quantitatively. Both
6 and
7 have copper atoms with a distorted square planar geometry. The averaged Cu(III)-C bonds of
6 and
7 (1.96–1.97 Å) are noticeably shorter than the Cu(I)-C bonds of
2–4. It is noteworthy that
6 and
7 represent a novel type of unprecedented organocopper(III) complex. By virtue of Cu L
2,3-edge X-ray absorption spectroscopy and experimentally calibrated electronic structure calculations, Lancaster and co-workers found that Cu(III) complex
7 has a LUMO (lowest unoccupied molecular orbital) with only ∼25% Cu 3d character, which suggests that the formal Cu(III) is not a physical d
8 configuration
[61].
Scheme 2. Organometallic spiro copper(I) and Cu(III) complexes
[59][60][59,60].
Figure 1. Molecular structures of
4,
5, and
7. Reprinted with permission from
[59][60][59,60]. Copyright 2017 and 2018 American Chemical Society.
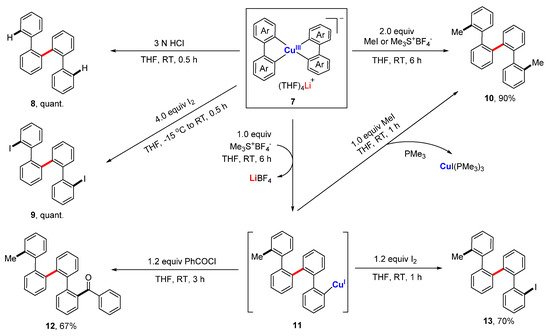
As revealed by the crystal structure of
7, all four aryl rings are at
cis-positions, which, in principle, provides a favorable arrangement for reductive elimination. However, no C-C bond formation occurred when the anionic compound
7 itself was refluxed overnight. However, when treated with electrophiles, e.g., aqueous HCl solution, iodine, and alkylation reagents,
7 afforded the symmetrical quaterphenyl derivatives
8–10 facilely in almost all quantitative yields via an electrophilic attack followed by a reduction elimination (
Scheme 3). A stepwise experiment showed that
7 reacted with 1.0 equiv. of methylation agent, producing a quaterphenylcopper(I) species
11, which can couple with other electrophiles to form the di-substituted quaterphenyl compounds
10,
12, and
13 in high yields. The reductive elimination of high-valent copper compounds has often been proposed as the final step to release the product, but there has been nearly no concrete experimental evidence of that until this work.
Scheme 3. Reductive elimination of organometallic spiro Cu(III) complexes.
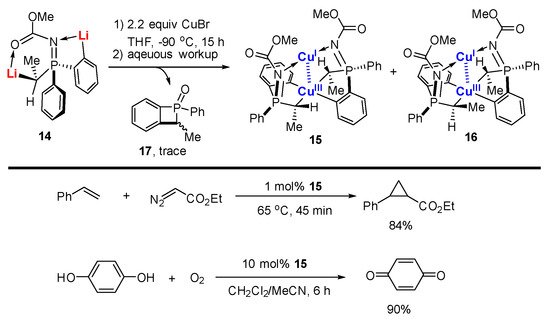
In a similar vein, López-Ortiz and co-workers reported the reaction between dilithiated phosphazene
14 with 2.2 equiv. of CuBr followed by an aqueous workup to afford two diastereotopic isomers of mixed-valent Cu(I)/Cu(III) complexes
15 and
16 in 53% combined yield, in which the Cu(III) center was chelated with two C,C-bidentate aryl-alkyl ligands (
Scheme 4)
[62]. A byproduct with a four-membered ring
17 was isolated as well. Both
15 and
16 can be purified by column chromatography. Accordingly, they are stable in air and moisture. Cu(III) and Cu(I) adopt a distorted square planar and linear geometry, respectively (
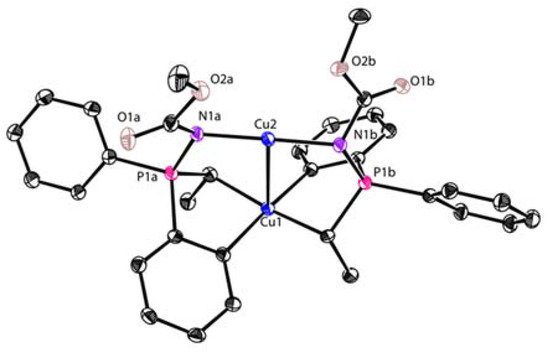
Figure 2). The averaged Cu(III)-C bond (1.98 Å) in
15 and
16 is similar to that in
6 and
7. The oxidation states of the Cu ions in
15 were further confirmed by extended X-ray absorption fine structure (EXAFS). Furthermore, all of the pieces of evidence, including the short Cu-Cu distance (2.74 Å) in the single-crystal structure, a critical bond point in the topological analysis of the X-ray experimental Fourier map as well as a diagnostic absorption band at 343 nm, strongly supported a weak metal–metal d
8-d
10 bonding between two Cu ions. Compound
15 can be used as a catalyst in catalytic cyclopropanation and aerobic oxidations. The characteristic signal of
15 has been observed and suggests that
15 remains unaffected during these transformations, probably due to its rigid structural configuration and extremely high stability.
Scheme 4. Mixed-valent bimetallic Cu(I)/Cu(III) compounds
[62].
Figure 2. Molecular structure of
15; drawn based on
[62].
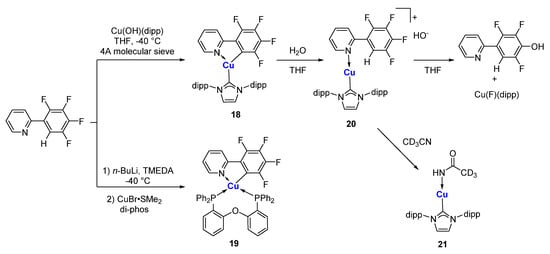
The compound 2-phenylpyridine and its derivatives are among the most popular scaffolds for photoactive transition metal complexes. Partial fluorination of the aryl ring makes the C-H bond at the 2-position very acidic. Additionally, at the same time, it increases the stability of the resulting Cu-C bond. Steffen and co-workers reported that the C,N-chelated organocopper compound
18 was obtained from a direct metalation from Cu(OH)(NHC), in which 4A molecular sieves play an essential role in removing water (
Scheme 5)
[63]. In the presence of water, compound
18 will inevitably undergo a hydrolysis process, regenerating the C-H bond and forming an ionic species
20. The counteranion of hydroxide in
20 will replace a fluoride at the 4-position via a nucleophilic aromatic substitution in THF to 4-hydroxyl-2-arylpyridine and copper fluoride species. When MeCN is used as the solvent, hydroxide will attack MeCN, forming a copper acetamide species
21. Similarly, a spiro complex
19 can also be prepared from lithiation and subsequent transmetalation with CuBr·SMe
2 in the presence of diphosphine ligands. It has been shown in single crystal structures that
18 and
19 adopt distorted trigonal and tetrahedral coordination geometries, respectively. In solution,
18 is weakly emissive. In the solid state, the C,N-chelated organocopper compounds exhibit intense orange-red luminescence (λ
max = 610 (
18), 607 (
19) nm) at room temperature and have phosphorescence lifetimes of τ = 8.6 (
18) and 9.5 (
19) μs.
Scheme 5. C,N-chelated organocopper compounds
[63].