 +1 credit
+1 credit
Video Upload Options
Cancer is a category of diseases involving abnormal cell growth with the potential to invade other parts of the body. Chemotherapy is the most widely used first-line treatment for multiple forms of cancer. Chemotherapeutic agents act via targeting the cellular apoptotic pathway. However, cancer cells usually acquire chemoresistance, leading to poor outcomes in cancer patients. For that reason, it is imperative to discover other cell death pathways for improved cancer intervention. Pyroptosis is a new form of programmed cell death that commonly occurs upon pathogen invasion. Pyroptosis is marked by cell swelling and plasma membrane rupture, which results in the release of cytosolic contents into the extracellular space. Currently, pyroptosis is proposed to be an alternative mode of cell death in cancer treatment. Accumulating evidence shows that the key components of pyroptotic cell death pathways, including inflammasomes, gasdermins and pro-inflammatory cytokines, are involved in the initiation and progression of cancer. Interfering with pyroptotic cell death pathways may represent a promising therapeutic option for cancer management.
1. Introduction
2. The Characteristics of Pyroptosis
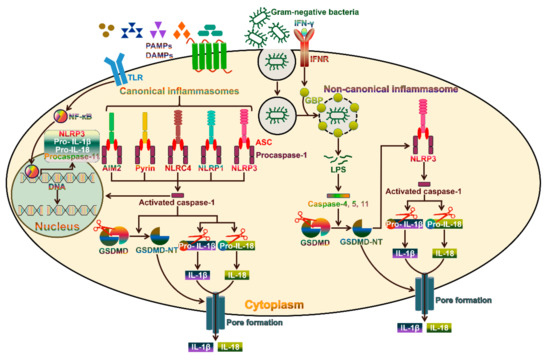
3. The Canonical Pyroptosis Pathway
4. The Emerging Role of Inflammasome-Dependent Cytokines in Cancer Pathogenesis
4.1. IL-1β and Cancer
4.2. Therapeutic Potential of IL-1 Neutralization in Cancer
4.3. IL-18 and Cancer
4.4. Potential Efficacy of Anti-IL-18 in Cancer Therapy
References
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host Cell Death and Inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109.
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell Biol. Int. 2019, 43, 582–592.
- He, Y.; Amer, A.O. Microbial Modulation of Host Apoptosis and Pyroptosis. Front. Cell Infect. Microbiol. 2014, 4, 83.
- Sharma, D.; Kanneganti, T.D. Inflammatory Cell Death in Intestinal Pathologies. Immunol. Rev. 2017, 280, 57–73.
- Fearnhead, H.O.; Rodriguez, J.; Govek, E.E.; Guo, W.; Kobayashi, R.; Hannon, G.; Lazebnik, Y.A. Oncogene-dependent Apoptosis is Mediated by Caspase-9. Proc. Natl. Acad. Sci. USA 1998, 95, 13664–13669.
- Ruhl, S.; Shkarina, K.; Demarco, B.; Heilig, R.; Santos, J.C.; Broz, P. ESCRT-dependent Membrane Repair Negatively Regulates Pyroptosis Downstream of GSDMD Activation. Science 2018, 362, 956–960.
- Weinlich, R.; Oberst, A.; Beere, H.M.; Green, D.R. Necroptosis in Development, Inflammation and Disease. Nat. Rev. Mol. Cell Biol. 2017, 18, 127–136.
- Dhuriya, Y.K.; Sharma, D. Necroptosis: A Regulated Inflammatory Mode of Cell Death. J. Neuroinflamm. 2018, 15, 199.
- Gong, Y.N.; Guy, C.; Crawford, J.C.; Green, D.R. Biological Events and Molecular Signaling following MLKL Activation during Necroptosis. Cell Cycle 2017, 16, 1748–1760.
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and Execution Mechanisms of Necroptosis: An Overview. Cell Death Differ. 2017, 24, 1184–1195.
- Gaidt, M.M.; Hornung, V. Pore Formation by GSDMD is the Effector Mechanism of Pyroptosis. EMBO J. 2016, 35, 2167–2169.
- Man, S.M.; Kanneganti, T.D. Gasdermin D: The Long-awaited Executioner of Pyroptosis. Cell Res. 2015, 25, 1183–1184.
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Xia, X.; Wang, X.; Zheng, Y.; Jiang, J.; Hu, J. What role does pyroptosis play in microbial infection? J. Cell. Physiol. 2019, 234, 7885–7892.
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75.
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103.
- Zhang, C.C.; Li, C.G.; Wang, Y.F.; Xu, L.H.; He, X.H.; Zeng, Q.Z.; Zeng, C.Y.; Mai, F.Y.; Hu, B.; Ouyang, D.Y. Chemotherapeutic paclitaxel and cisplatin differentially induce pyroptosis in A549 lung cancer cells via caspase-3/GSDME activation. Apoptosis 2019, 24, 312–325.
- Bauernfeind, F.; Hornung, V. Of inflammasomes and pathogens—Sensing of microbes by the inflammasome. EMBO Mol. Med. 2013, 5, 814–826.
- Stowe, I.; Lee, B.; Kayagaki, N. Caspase-11: Arming the guards against bacterial infection. Immunol. Rev. 2015, 265, 75–84.
- Fernandes-Alnemri, T.; Wu, J.; Yu, J.W.; Datta, P.; Miller, B.; Jankowski, W.; Rosenberg, S.; Zhang, J.; Alnemri, E.S. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 2007, 14, 1590–1604.
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832.
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629.
- Rubartelli, A.; Lotze, M.T. Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol. 2007, 28, 429–436.
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735.
- Sharma, N.; Jha, S. NLR-regulated pathways in cancer: Opportunities and obstacles for therapeutic interventions. Cell. Mol. Life Sci. CMLS 2016, 73, 1741–1764.
- Awad, F.; Assrawi, E.; Jumeau, C.; Georgin-Lavialle, S.; Cobret, L.; Duquesnoy, P.; Piterboth, W.; Thomas, L.; Stankovic-Stojanovic, K.; Louvrier, C.; et al. Impact of human monocyte and macrophage polarization on NLR expression and NLRP3 inflammasome activation. PLoS ONE 2017, 12, e0175336.
- Caneparo, V.; Landolfo, S.; Gariglio, M.; De Andrea, M. The Absent in Melanoma 2-Like Receptor IFN-Inducible Protein 16 as an Inflammasome Regulator in Systemic Lupus Erythematosus: The Dark Side of Sensing Microbes. Front. Immunol. 2018, 9, 1180.
- Man, S.M.; Karki, R.; Kanneganti, T.D. AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur. J. Immunol. 2016, 46, 269–280.
- Lee, J.; Li, L.; Gretz, N.; Gebert, J.; Dihlmann, S. Absent in Melanoma 2 (AIM2) is an important mediator of interferon-dependent and -independent HLA-DRA and HLA-DRB gene expression in colorectal cancers. Oncogene 2012, 31, 1242–1253.
- Yu, J.W.; Wu, J.; Zhang, Z.; Datta, P.; Ibrahimi, I.; Taniguchi, S.; Sagara, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Cryopyrin and pyrin activate caspase-1, but not NF-kappaB, via ASC oligomerization. Cell Death Differ. 2006, 13, 236–249.
- De Torre-Minguela, C.; Mesa Del Castillo, P.; Pelegrin, P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front. Immunol. 2017, 8, 43.
- Garib, F.Y.; Rizopulu, A.P.; Kuchmiy, A.A.; Garib, V.F. Inactivation of Inflammasomes by Pathogens Regulates Inflammation. Biochem. Biokhimiia 2016, 81, 1326–1339.
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog. 2012, 8, e1002638.
- Hellmich, K.A.; Levinsohn, J.L.; Fattah, R.; Newman, Z.L.; Maier, N.; Sastalla, I.; Liu, S.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleaves mouse nlrp1b in both toxin-sensitive and toxin-resistant macrophages. PLoS ONE 2012, 7, e49741.
- Qu, Y.; Misaghi, S.; Izrael-Tomasevic, A.; Newton, K.; Gilmour, L.L.; Lamkanfi, M.; Louie, S.; Kayagaki, N.; Liu, J.; Komuves, L.; et al. Phosphorylation of NLRC4 is critical for inflammasome activation. Nature 2012, 490, 539–542.
- Morrone, S.R.; Matyszewski, M.; Yu, X.; Delannoy, M.; Egelman, E.H.; Sohn, J. Assembly-driven activation of the AIM2 foreign-dsDNA sensor provides a polymerization template for downstream ASC. Nat. Commun. 2015, 6, 7827.
- Jamilloux, Y.; Magnotti, F.; Belot, A.; Henry, T. The pyrin inflammasome: from sensing RhoA GTPases-inhibiting toxins to triggering autoinflammatory syndromes. Pathog. Dis. 2018, 76, fty020.
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206.
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for caspase-1. J. Biol. Chem. 2002, 277, 21119–21122.
- Malik, A.; Kanneganti, T.D. Inflammasome activation and assembly at a glance. J. Cell Sci. 2017, 130, 3955–3963.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116.
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158.
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684.
- Ruan, J.; Xia, S.; Liu, X.; Lieberman, J.; Wu, H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 2018, 557, 62–67.
- Kepp, O.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Pyroptosis—A cell death modality of its kind? Eur. J. Immunol. 2010, 40, 627–630.
- Banerjee, I.; Behl, B.; Mendonca, M.; Shrivastava, G.; Russo, A.J.; Menoret, A.; Ghosh, A.; Vella, A.T.; Vanaja, S.K.; Sarkar, S.N.; et al. Gasdermin D Restrains Type I Interferon Response to Cytosolic DNA by Disrupting Ionic Homeostasis. Immunity 2018, 49, 413–426.e5.
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153.
- Ruhl, S.; Broz, P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur. J. Immunol. 2015, 45, 2927–2936.
- Monteleone, M.; Stow, J.L.; Schroder, K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine 2015, 74, 213–218.
- Monteleone, M.; Stanley, A.C.; Chen, K.W.; Brown, D.L.; Bezbradica, J.S.; von Pein, J.B.; Holley, C.L.; Boucher, D.; Shakespear, M.R.; Kapetanovic, R.; et al. Interleukin-1beta Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep. 2018, 24, 1425–1433.
- Gutierrez, K.D.; Davis, M.A.; Daniels, B.P.; Olsen, T.M.; Ralli-Jain, P.; Tait, S.W.; Gale, M., Jr.; Oberst, A. MLKL Activation Triggers NLRP3-Mediated Processing and Release of IL-1beta Independently of Gasdermin-D. J. Immunol. 2017, 198, 2156–2164.
- Lage, S.L.; Dominical, V.M.; Wong, C.S.; Sereti, I. Evaluation of Canonical Inflammasome Activation in Human Monocytes by Imaging Flow Cytometry. Front. Immunol. 2019, 10, 1284.
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. CB 2016, 26, R568–R572.
- Van Gorp, H.; Lamkanfi, M. The emerging roles of inflammasome-dependent cytokines in cancer development. EMBO Rep. 2019, 20.
- Dinarello, C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 2011, 117, 3720–3732.
- Boraschi, D.; Italiani, P.; Weil, S.; Martin, M.U. The family of the interleukin-1 receptors. Immunol. Rev. 2018, 281, 197–232.
- Munoz-Wolf, N.; Lavelle, E.C. A Guide to IL-1 family cytokines in adjuvanticity. FEBS J. 2018, 285, 2377–2401.
- Kaplanov, I.; Carmi, Y.; Kornetsky, R.; Shemesh, A.; Shurin, G.V.; Shurin, M.R.; Dinarello, C.A.; Voronov, E.; Apte, R.N. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. USA 2019, 116, 1361–1369.
- Wu, J.; Hong, Y.; Wu, T.; Wang, J.; Chen, X.; Wang, Z.; Cheng, B.; Xia, J. Stromal-epithelial lactate shuttle induced by tumorderived interleukin1beta promotes cell proliferation in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 41, 687–696.
- Hu, M.; Yuan, X.; Liu, Y.; Tang, S.; Miao, J.; Zhou, Q.; Chen, S. IL-1beta-induced NF-kappaB activation down-regulates miR-506 expression to promotes osteosarcoma cell growth through JAG1. Biomed. Pharmacother. 2017, 95, 1147–1155.
- Liu, B.; Zhou, Y.; Chen, X.; Peng, D. IL-1beta-mediated NF-kappaB signaling augments the osteosarcoma cell growth through modulating miR-376c/TGFA axis. Die Pharm. 2017, 72, 419–424.
- Yu, A.; Wang, Y.; Bian, Y.; Chen, L.; Guo, J.; Shen, W.; Chen, D.; Liu, S.; Sun, X. IL-1beta promotes the nuclear translocaiton of S100A4 protein in gastric cancer cells MGC803 and the cell’s stem-like properties through PI3K pathway. J. Cell. Biochem. 2018, 119, 8163–8173.
- Wang, X.; Wang, B.; Xie, J.; Hou, D.; Zhang, H.; Huang, H. Melatonin inhibits epithelialtomesenchymal transition in gastric cancer cells via attenuation of IL1beta/NFkappaB/MMP2/MMP9 signaling. Int. J. Mol. Med. 2018, 42, 2221–2228.
- Li, T.; Zhu, J.; Zuo, S.; Chen, S.; Ma, J.; Ma, Y.; Guo, S.; Wang, P.; Liu, Y. 1,25(OH)2D3 attenuates IL-1beta-induced epithelial to mesenchymal transition through inhibiting the expression of lncTCF7. Oncol. Res. 2018, 27, 739–750.
- Castano, Z.; San Juan, B.P.; Spiegel, A.; Pant, A.; DeCristo, M.J.; Laszewski, T.; Ubellacker, J.M.; Janssen, S.R.; Dongre, A.; Reinhardt, F.; et al. IL-1beta inflammatory response driven by primary breast cancer prevents metastasis-initiating cell colonization. Nat. Cell Biol. 2018, 20, 1084–1097.
- Jimenez-Garduno, A.M.; Mendoza-Rodriguez, M.G.; Urrutia-Cabrera, D.; Dominguez-Robles, M.C.; Perez-Yepez, E.A.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induced methylation of the estrogen receptor ERalpha gene correlates with EMT and chemoresistance in breast cancer cells. Biochem. Biophys. Res. Commun. 2017, 490, 780–785.
- Storr, S.J.; Safuan, S.; Ahmad, N.; El-Refaee, M.; Jackson, A.M.; Martin, S.G. Macrophage-derived interleukin-1beta promotes human breast cancer cell migration and lymphatic adhesion in vitro. Cancer Immunol. Immunother. CII 2017, 66, 1287–1294.
- Mon, N.N.; Senga, T.; Ito, S. Interleukin-1beta activates focal adhesion kinase and Src to induce matrix metalloproteinase-9 production and invasion of MCF-7 breast cancer cells. Oncol. Lett. 2017, 13, 955–960.
- Zhou, J.; Zheng, S.; Liu, T.; Liu, Q.; Chen, Y.; Tan, D.; Ma, R.; Lu, X. IL-1beta from M2 macrophages promotes migration and invasion of ESCC cells enhancing epithelial-mesenchymal transition and activating NF-kappaB signaling pathway. J. Cell. Biochem. 2018, 119, 7040–7052.
- Zhang, J.; Zhang, Q.; Lou, Y.; Fu, Q.; Chen, Q.; Wei, T.; Yang, J.; Tang, J.; Wang, J.; Chen, Y.; et al. Hypoxia-inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology 2018, 67, 1872–1889.
- Wang, C.I.; Yu, C.J.; Huang, Y.; Yi, J.S.; Cheng, H.W.; Kao, H.K.; Lao, W.W.; Chang, K.P. Association of overexpressed karyopherin alpha 2 with poor survival and its contribution to interleukin-1beta-induced matrix metalloproteinase expression in oral cancer. Head Neck 2018, 40, 1719–1733.
- Lee, M.K.; Park, J.H.; Gi, S.H.; Hwang, Y.S. IL-1beta Induces Fascin Expression and Increases Cancer Invasion. Anticancer Res. 2018, 38, 6127–6132.
- Chen, X.; Lv, Q.; Hong, Y.; Chen, X.; Cheng, B.; Wu, T. IL-1beta maintains the redox balance by regulating glutaredoxin 1 expression during oral carcinogenesis. J. Oral Pathol. Med. 2017, 46, 332–339.
- Mendoza-Rodriguez, M.G.; Ayala-Sumuano, J.T.; Garcia-Morales, L.; Zamudio-Meza, H.; Perez-Yepez, E.A.; Meza, I. IL-1beta Inflammatory Cytokine-Induced TP63 Isoform NP63alpha Signaling Cascade Contributes to Cisplatin Resistance in Human Breast Cancer Cells. Int. J. Mol. Sci. 2019, 20, 270.
- Mendoza-Rodriguez, M.; Arevalo Romero, H.; Fuentes-Panana, E.M.; Ayala-Sumuano, J.T.; Meza, I. IL-1beta induces up-regulation of BIRC3, a gene involved in chemoresistance to doxorubicin in breast cancer cells. Cancer Lett. 2017, 390, 39–44.
- Zhang, D.; Li, L.; Jiang, H.; Li, Q.; Wang-Gillam, A.; Yu, J.; Head, R.; Liu, J.; Ruzinova, M.B.; Lim, K.H. Tumor-Stroma IL1beta-IRAK4 Feedforward Circuitry Drives Tumor Fibrosis, Chemoresistance, and Poor Prognosis in Pancreatic Cancer. Cancer Res. 2018, 78, 1700–1712.
- Dinarello, C.A. An expanding role for interleukin-1 blockade from gout to cancer. Mol. Med. 2014, 20 (Suppl. 1), S43–S58.
- Braddock, M.; Quinn, A. Targeting IL-1 in inflammatory disease: New opportunities for therapeutic intervention. Nat. Rev. Drug Discov. 2004, 3, 330–339.
- Fleischmann, R.; Stern, R.; Iqbal, I. Anakinra: an inhibitor of IL-1 for the treatment of rheumatoid arthritis. Expert Opin. Biol. Ther. 2004, 4, 1333–1344.
- Zhang, B.; Chu, S.; Agarwal, P.; Campbell, V.L.; Hopcroft, L.; Jorgensen, H.G.; Lin, A.; Gaal, K.; Holyoake, T.L.; Bhatia, R. Inhibition of interleukin-1 signaling enhances elimination of tyrosine kinase inhibitor-treated CML stem cells. Blood 2016, 128, 2671–2682.
- Kurzrock, R.; Hickish, T.; Wyrwicz, L.; Saunders, M.; Wu, Q.; Stecher, M.; Mohanty, P.; Dinarello, C.A.; Simard, J. Interleukin-1 receptor antagonist levels predict favorable outcome after bermekimab, a first-in-class true human interleukin-1alpha antibody, in a phase III randomized study of advanced colorectal cancer. Oncoimmunology 2019, 8, 1551651.
- Stanam, A.; Gibson-Corley, K.N.; Love-Homan, L.; Ihejirika, N.; Simons, A.L. Interleukin-1 blockade overcomes erlotinib resistance in head and neck squamous cell carcinoma. Oncotarget 2016, 7, 76087–76100.
- Isambert, N.; Hervieu, A.; Rebe, C.; Hennequin, A.; Borg, C.; Zanetta, S.; Chevriaux, A.; Richard, C.; Derangere, V.; Limagne, E.; et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): A single-arm phase 2 study. Oncoimmunology 2018, 7, e1474319.
- Zhuang, Z.; Ju, H.Q.; Aguilar, M.; Gocho, T.; Li, H.; Iida, T.; Lee, H.; Fan, X.; Zhou, H.; Ling, J.; et al. IL1 Receptor Antagonist Inhibits Pancreatic Cancer Growth by Abrogating NF-kappaB Activation. Clin. Cancer Res. 2016, 22, 1432–1444.
- Church, L.D.; McDermott, M.F. Canakinumab, a fully-human mAb against IL-1beta for the potential treatment of inflammatory disorders. Curr. Opin. Mol. Ther. 2009, 11, 81–89.
- Tulotta, C.; Lefley, D.V.; Freeman, K.; Gregory, W.M.; Hanby, A.M.; Heath, P.R.; Nutter, F.; Wilkinson, J.M.; Spicer-Hadlington, A.R.; Liu, X.; et al. Endogenous Production of IL1B by Breast Cancer Cells Drives Metastasis and Colonization of the Bone Microenvironment. Clin. Cancer Res. 2019, 25, 2769–2782.
- Orlando, I.; Vitale, A.; Rigante, D.; Lopalco, G.; Fabiani, C.; Cantarini, L. Long-term efficacy and safety of the interleukin-1 inhibitors anakinra and canakinumab in refractory Behcet disease uveitis and concomitant bladder papillary carcinoma. Intern. Med. J. 2017, 47, 1086–1088.
- Thompson, P.L.; Nidorf, S.M. Anti-inflammatory therapy with canakinumab for atherosclerotic disease: Lessons from the CANTOS trial. J. Thorac. Dis. 2018, 10, 695–698.
- Ridker, P.M.; MacFadyen, J.G.; Thuren, T.; Everett, B.M.; Libby, P.; Glynn, R.J.; Group, C.T. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: Exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 2017, 390, 1833–1842.
- Dinarello, C.A.; Simon, A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012, 11, 633–652.
- Garg, M.; de Jesus, A.A.; Chapelle, D.; Dancey, P.; Herzog, R.; Rivas-Chacon, R.; Muskardin, T.L.W.; Reed, A.; Reynolds, J.C.; Goldbach-Mansky, R.; et al. Rilonacept maintains long-term inflammatory remission in patients with deficiency of the IL-1 receptor antagonist. JCI Insight 2017, 2, e94838.
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484.
- Wawrocki, S.; Druszczynska, M.; Kowalewicz-Kulbat, M.; Rudnicka, W. Interleukin 18 (IL-18) as a target for immune intervention. Acta Biochim. Pol. 2016, 63, 59–63.
- Enoksson, S.L.; Grasset, E.K.; Hagglof, T.; Mattsson, N.; Kaiser, Y.; Gabrielsson, S.; McGaha, T.L.; Scheynius, A.; Karlsson, M.C. The inflammatory cytokine IL-18 induces self-reactive innate antibody responses regulated by natural killer T cells. Proc. Natl. Acad. Sci. USA 2011, 108, E1399–E1407.
- Sedimbi, S.K.; Hagglof, T.; Karlsson, M.C. IL-18 in inflammatory and autoimmune disease. Cell. Mol. Life Sci. CMLS 2013, 70, 4795–4808.
- Jurecekova, J.; Babusikova, E.; Kmetova Sivonova, M.; Drobkova, H.; Petras, M.; Kliment, J.; Halasova, E. Association between interleukin-18 variants and prostate cancer in Slovak population. Neoplasma 2017, 64, 148–155.
- Huang, C.Y.; Chang, W.S.; Tsai, C.W.; Hsia, T.C.; Shen, T.C.; Bau, D.T.; Shui, H.A. Interleukin-18 promoter genotype is associated with the risk of nasopharyngeal carcinoma in Taiwan. Cancer Manag. Res. 2018, 10, 5199–5207.
- Bakr, N.M.; Awad, A.; Moustafa, E.A. Association of genetic variants in the interleukin-18 gene promoter with risk of hepatocellular carcinoma and metastasis in patients with hepatitis C virus infection. IUBMB Life 2018, 70, 165–174.
- Gan, W.Y.; Li, H.M.; Zhang, Y.G.; Li, C.M.; Wang, Y. Association between IL18-607C/A and -137G/C polymorphisms and susceptibility to non-small cell lung cancer in a Chinese population. Genet. Mol. Res. GMR 2016, 15, gmr15048822.
- Wang, H.; Hua, M.; Wang, S.; Yu, J.; Chen, C.; Zhao, X.; Zhang, C.; Zhong, C.; Wang, R.; He, N.; et al. Genetic polymorphisms of IL-18 rs1946518 and IL-1beta rs16944 are associated with prognosis and survival of acute myeloid leukemia. Inflamm. Res. 2017, 66, 249–258.
- Nakamura, K.; Kassem, S.; Cleynen, A.; Chretien, M.L.; Guillerey, C.; Putz, E.M.; Bald, T.; Forster, I.; Vuckovic, S.; Hill, G.R.; et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 2018, 33, 634–648.e5.
- Ko, C.Y.; Wang, W.L.; Li, C.F.; Jeng, Y.M.; Chu, Y.Y.; Wang, H.Y.; Tseng, J.T.; Wang, J.M. IL-18-induced interaction between IMP3 and HuR contributes to COX-2 mRNA stabilization in acute myeloid leukemia. J. Leukoc. Biol. 2016, 99, 131–141.
- Ding, S.; Tang, Z.; Jiang, Y.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. IL-8 Is Involved in Estrogen-Related Receptor alpha-Regulated Proliferation and Migration of Colorectal Cancer Cells. Dig. Dis. Sci. 2017, 62, 3438–3446.
- Li, Y.; Xu, Z.; Li, J.; Ban, S.; Duan, C.; Liu, W. Interleukin-18 expression in oral squamous cell carcinoma: its role in tumor cell migration and invasion, and growth of tumor cell xenografts. FEBS Open Bio 2018, 8, 1953–1963.
- Usul Afsar, C.; Karabulut, M.; Karabulut, S.; Alis, H.; Gonenc, M.; Dagoglu, N.; Serilmez, M.; Tas, F. Circulating interleukin-18 (IL-18) is a predictor of response to gemcitabine based chemotherapy in patients with pancreatic adenocarcinoma. J. Infect. Chemother. 2017, 23, 196–200.
- Fabbi, M.; Carbotti, G.; Ferrini, S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J. Leukoc. Biol. 2015, 97, 665–675.
- Kaplanski, G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol. Rev. 2018, 281, 138–153.
- Li, J.; Qiu, G.; Fang, B.; Dai, X.; Cai, J. Deficiency of IL-18 Aggravates Esophageal Carcinoma Through Inhibiting IFN-gamma Production by CD8(+)T Cells and NK Cells. Inflammation 2018, 41, 667–676.
- Xu, X.; Song, C.; Chen, Z.; Yu, C.; Wang, Y.; Tang, Y.; Luo, J. Downregulation of HuR Inhibits the Progression of Esophageal Cancer through Interleukin-18. Cancer Res. Treat. 2018, 50, 71–87.
- Senju, H.; Kumagai, A.; Nakamura, Y.; Yamaguchi, H.; Nakatomi, K.; Fukami, S.; Shiraishi, K.; Harada, Y.; Nakamura, M.; Okamura, H.; et al. Effect of IL-18 on the Expansion and Phenotype of Human Natural Killer Cells: Application to Cancer Immunotherapy. Int. J. Biol. Sci. 2018, 14, 331–340.
- Kunert, A.; Chmielewski, M.; Wijers, R.; Berrevoets, C.; Abken, H.; Debets, R. Intra-tumoral production of IL18, but not IL12, by TCR-engineered T cells is non-toxic and counteracts immune evasion of solid tumors. Oncoimmunology 2017, 7, e1378842.
- Yang, C.; Cao, H.; Liu, N.; Xu, K.; Ding, M.; Mao, L.J. Oncolytic adenovirus expressing interleukin-18 improves antitumor activity of dacarbazine for malignant melanoma. Drug Des. Dev. Ther. 2016, 10, 3755–3761.
- Markowitz, G.J.; Yang, P.; Fu, J.; Michelotti, G.A.; Chen, R.; Sui, J.; Yang, B.; Qin, W.H.; Zhang, Z.; Wang, F.S.; et al. Inflammation-Dependent IL18 Signaling Restricts Hepatocellular Carcinoma Growth by Enhancing the Accumulation and Activity of Tumor-Infiltrating Lymphocytes. Cancer Res. 2016, 76, 2394–2405.
- Zhuang, L.; Fulton, R.J.; Rettman, P.; Sayan, A.E.; Coad, J.; Al-Shamkhani, A.; Khakoo, S.I. Activity of IL-12/15/18 primed natural killer cells against hepatocellular carcinoma. Hepatol. Int. 2019, 13, 75–83.
- Liu, X.; Hu, J.; Li, Y.; Cao, W.; Wang, Y.; Ma, Z.; Li, F. Mesenchymal stem cells expressing interleukin-18 inhibit breast cancer in a mouse model. Oncol. Lett. 2018, 15, 6265–6274.
- Zhao, Y.; Shen, M.; Feng, Y.; He, R.; Xu, X.; Xie, Y.; Shi, X.; Zhou, M.; Pan, S.; Wang, M.; et al. Regulatory B cells induced by pancreatic cancer cell-derived interleukin-18 promote immune tolerance via the PD-1/PD-L1 pathway. Oncotarget 2018, 9, 14803–14814.
- Guo, X.; Zheng, L.; Jiang, J.; Zhao, Y.; Wang, X.; Shen, M.; Zhu, F.; Tian, R.; Shi, C.; Xu, M.; et al. Blocking NF-kappaB Is Essential for the Immunotherapeutic Effect of Recombinant IL18 in Pancreatic Cancer. Clin. Cancer Res. 2016, 22, 5939–5950.
- Park, I.H.; Yang, H.N.; Lee, K.J.; Kim, T.S.; Lee, E.S.; Jung, S.Y.; Kwon, Y.; Kong, S.Y. Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 2017, 8, 32722–32730.
- Li, K.; Wei, L.; Huang, Y.; Wu, Y.; Su, M.; Pang, X.; Wang, N.; Ji, F.; Zhong, C.; Chen, T. Leptin promotes breast cancer cell migration and invasion via IL-18 expression and secretion. Int. J. Oncol. 2016, 48, 2479–2487.
- Ma, Z.; Li, W.; Yoshiya, S.; Xu, Y.; Hata, M.; El-Darawish, Y.; Markova, T.; Yamanishi, K.; Yamanishi, H.; Tahara, H.; et al. Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin. Cancer Res. 2016, 22, 2969–2980.
- Robertson, M.J.; Kirkwood, J.M.; Logan, T.F.; Koch, K.M.; Kathman, S.; Kirby, L.C.; Bell, W.N.; Thurmond, L.M.; Weisenbach, J.; Dar, M.M. A dose-escalation study of recombinant human interleukin-18 using two different schedules of administration in patients with cancer. Clin. Cancer Res. 2008, 14, 3462–3469.
- Robertson, M.J.; Mier, J.W.; Logan, T.; Atkins, M.; Koon, H.; Koch, K.M.; Kathman, S.; Pandite, L.N.; Oei, C.; Kirby, L.C.; et al. Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin. Cancer Res. 2006, 12, 4265–4273.
- Tarhini, A.A.; Millward, M.; Mainwaring, P.; Kefford, R.; Logan, T.; Pavlick, A.; Kathman, S.J.; Laubscher, K.H.; Dar, M.M.; Kirkwood, J.M. A phase 2, randomized study of SB-485232, rhIL-18, in patients with previously untreated metastatic melanoma. Cancer 2009, 115, 859–868.
- Robertson, M.J.; Kline, J.; Struemper, H.; Koch, K.M.; Bauman, J.W.; Gardner, O.S.; Murray, S.C.; Germaschewski, F.; Weisenbach, J.; Jonak, Z.; et al. A dose-escalation study of recombinant human interleukin-18 in combination with rituximab in patients with non-Hodgkin lymphoma. J. Immunother. 2013, 36, 331–341.
- Simpkins, F.; Flores, A.; Chu, C.; Berek, J.S.; Lucci, J., 3rd; Murray, S.; Bauman, J.; Struemper, H.; Germaschewski, F.; Jonak, Z.; et al. Chemoimmunotherapy using pegylated liposomal Doxorubicin and interleukin-18 in recurrent ovarian cancer: a phase I dose-escalation study. Cancer Immunol. Res. 2013, 1, 168–178.
- Cao, Q.; Cai, W.; Niu, G.; He, L.; Chen, X. Multimodality imaging of IL-18–binding protein-Fc therapy of experimental lung metastasis. Clin. Cancer Res. 2008, 14, 6137–6145.

