Cancer is a category of diseases involving abnormal cell growth with the potential to invade other parts of the body. Chemotherapy is the most widely used first-line treatment for multiple forms of cancer. Chemotherapeutic agents act via targeting the cellular apoptotic pathway. However, cancer cells usually acquire chemoresistance, leading to poor outcomes in cancer patients. For that reason, it is imperative to discover other cell death pathways for improved cancer intervention. Pyroptosis is a new form of programmed cell death that commonly occurs upon pathogen invasion. Pyroptosis is marked by cell swelling and plasma membrane rupture, which results in the release of cytosolic contents into the extracellular space. Currently, pyroptosis is proposed to be an alternative mode of cell death in cancer treatment. Accumulating evidence shows that the key components of pyroptotic cell death pathways, including inflammasomes, gasdermins and pro-inflammatory cytokines, are involved in the initiation and progression of cancer. Interfering with pyroptotic cell death pathways may represent a promising therapeutic option for cancer management.
1. Introduction
Cell death is a crucial phenomenon in biological activities and serves a pivotal role in maintaining homeostatic balance in vivo [
1]. At present, several forms of cell death, such as apoptosis, necroptosis and pyroptosis, have been found [
2]. As a form of non-inflammatory programmed cell death, apoptosis can be induced by either intrinsic or extrinsic factors, followed by sequential activation of initiator and executioner caspases [
3,
4]. Both apoptosis and pyroptosis are executed by caspases. Apoptosis is mediated by caspase-2, -3, -6, -7, -8 and -9 [
5]. The term pyroptosis was initially proposed in 2001. Pyroptosis is inherently a type of programmed cell death that is initiated by inflammatory caspases (caspase-1, -4, -5 and -11) upon activation of the canonical or non-canonical inflammasome pathways [
6]. The requirement of inflammatory caspases in executing pyroptosis differentiates it from another inflammatory and necrotic form of programmed cell death known as necroptosis [
7]. Necroptosis is a form of regulated cell death mediated by receptor-interacting serine/threonine-protein kinase 3 (RIPK3) (all the abbreviations are listed in
Table S1) and its downstream substrate mixed lineage kinase domain-like pseudokinase (MLKL) [
8]. RIPK3 phosphorylates the necroptosis executioner MLKL, resulting in the formation of MLKL oligomers, which then translocate to the plasma membrane [
9]. These events ultimately lead to necrotic plasma membrane permeabilization and cell death associated with loss of cell and organelle integrity [
10]. Necroptosis is widely known as a defense mechanism against viral infection and can induce programmed cell death in virus-infected cells.
Recently, pyroptosis has become a research hotspot in programmed cell death. The induction of pyroptosis requires the activation of the pore-forming protein gasdermin D (GSDMD) by inflammatory caspases [
11]. In the canonical inflammasome pathway, caspase-1 mediates the cleavage of GSDMD and the maturation of pro-inflammatory cytokines, interleukin-1β (IL-1β) and interleukin-18 (IL-18) [
12]. GSDMD pores favor the leakage of intracellular components into the extracellular environment. Unlike canonical inflammasomes, the non-canonical inflammasome pathway can be initiated by the direct binding of caspase-4, -5 and -11 to lipopolysaccharide (LPS) from Gram-negative bacteria [
13]. These caspases activate GSDMD to induce cell lysis and death. In addition, caspase-1 is activated in the non-classical pyroptosis pathway, leading to the production of IL-1β and IL-18, which are liberated into the extracellular milieu [
14]. Pyroptosis serves a vital role in host defense response against pathogens [
15]. Pyroptotic cell death, started by pathogen infection, contributes to the release of cytosolic contents from infected cells, hence triggering an inflammatory cascade [
16]. Local inflammation results in recruitment and activation of immune cells, ultimately facilitating the clearance of invading pathogens. Of note, recent studies have confirmed that pyroptosis functions in orchestrating cancer cell death [
17,
18]. Moreover, inflammasomes, gasdermins and pro-inflammatory cytokines can regulate key processes involved in cancer development. Therefore, pyroptotic cell death pathways constitute a novel mechanism contributing to cancer pathogenesis.
2. The Characteristics of Pyroptosis
Pyroptosis, a pro-inflammatory form of cell death, is generally induced by intracellular pathogen infection and forms part of the host defense system [
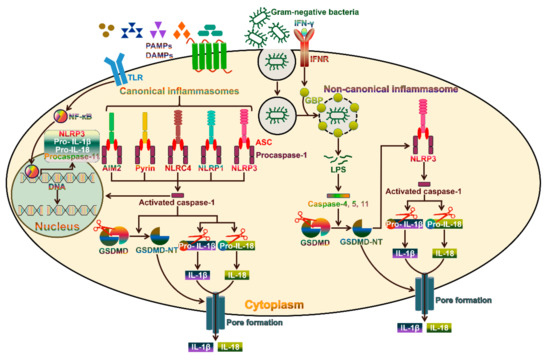
19]. Pyroptosis can be induced via two pathways, the canonical and non-canonical inflammasome pathways (
Figure 1). Canonical pyroptosis is executed by caspase-1, which is triggered by a number of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), while non-canonical pyroptosis is dependent on human caspase-4/-5 or mouse caspase-11 and can be induced by intracellular LPS [
20]. The morphological features of caspase-1-dependent pyroptosis and caspase-1-independent pyroptosis are similar. Both are characterized by the loss of cellular membrane integrity, chromatin condensation and DNA fragmentation. Specially, the cellular membrane undergoes rupture, resealing and swelling, and forms a balloon-shaped vesicle around the nucleus [
21]. The cellular membrane becomes disrupted, and the cytoplasmic materials, including pro-inflammatory cytokines, endogenous DAMPs and alarmins, are released into the extracellular space [
16].
Figure 1. Molecular mechanisms involved in inflammasome activation. Activation of the canonical inflammasomes usually requires two steps. As the first priming step, PAMPs or DAMPs combine with TLRs on the plasma membrane to trigger NF-ĸB-dependent transcription of inflammasome components and their downstream targets, including NLRP3, pro-IL-1β and pro-IL-18. The second step involves the recognition of PAMPs or DAMPs by the inflammasome sensors AIM2, pyrin, NLRC4, NLRP1 and NLRP3. These sensors recruit the adaptor protein ASC and procaspase-1 to form diverse inflammasomes. Once assembled, the inflammasomes act as activating platforms for procaspase-1. Catalytically active caspase-1 converts the inactive pro-IL-1β and pro-IL-18 into their biologically active forms (IL-1β and IL-18). On the other hand, caspase-1 cleaves GSDMD to release an N-terminal pore-forming domain (GSDMD-NT), which migrates toward the plasma membrane and forms membrane pores. The pores facilitate the release of the active IL-1β and IL-18 into the extracellular environment. In addition, caspase-1 activation leads to the cleavage of chromosomal DNA by nuclease activity. The non-canonical inflammasome pathway is specifically activated by Gram-negative bacteria. IFN-γ-induced GBP causes the lysis of Gram-negative bacteria-containing vacuoles, allowing the release of LPS into the cytoplasm. Intracellular bacterial LPS can directly activate caspase-4 and -5 in humans and caspase-11 in mice. These caspases cleave GSDMD to drive pyroptotic cell death. GSDMD-NT activates the NLRP3 inflammasome to promote caspase-1-dependent cytokine maturation. TLR, toll-like receptor; PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; IFN-γ, interferon-γ; IFNR, interferon receptor; NF-ĸB, nuclear factor-ĸB; GBP, guanylate-binding protein; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; LPS, lipopolysaccharide; GSDMD, gasdermin D; GSDMD-NT, the N-terminal fragment of GSDMD; IL-1β, interleukin-1β; IL-18, interleukin-18.
3. The Canonical Pyroptosis Pathway
Inflammasomes are macromolecular protein complexes consisting of an inflammasome sensor, the adaptor protein apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) and caspase-1 [
22]. Based on their structural features, the inflammasome sensors are categorized into nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), absent in melanoma 2 (AIM2)-like receptors (ALRs) and pyrin [
23]. The NLRs belong to host pattern recognition receptors (PRRs) and act as intracellular sensors of PAMPs and DAMPs [
24]. The NLRs are composed of three main domains: An N-terminal caspase recruitment domain (CARD) or pyrin domain, a middle nucleotide-binding domain (NBD or NACHT) and a C-terminal leucine-rich repeat (LRR) domain [
25]. The NBD domain mediates NLR oligomerization, while the LRR acts as a sensor of PAMPs and DAMPs [
26]. Once formed, the N-terminal pyrin or CARD domain of NLR oligomers recruits and seeds ASC or caspase-1. Among NLRs, NLR family pyrin domain-containing 1 (NLRP1), NLRP2, NLRP3, NLRP6, NLRP7, NLRP12 and NLR family CARD domain-containing protein 4 (NLRC4) are regarded as inflammasome-nucleating proteins [
27]. The ALRs are a newly characterized class of PRRs that can detect pathogen DNAs in both the cytosol and nucleus [
28]. The ALR family member AIM2 consists of an N-terminal pyrin domain, which binds to the pyrin domain of ASC, and a C-terminal hematopoietic interferon (IFN)-inducible nuclear protein containing a 200-amino-acid repeat (HIN-200) domain, which directly interacts with dsDNA [
29]. AIM2 can initiate inflammasome assembly upon activation leading to caspase-1-mediated inflammatory responses and cell death [
30]. Pyrin is another vital inflammasome-forming protein [
31]. Pyrin harbors an N-terminal pyrin domain that is responsible for its combination with ASC and subsequent activation of caspase-1 [
32].
Five canonical inflammasomes, NLRP1, NLRP3, NLRC4, AIM2 and pyrin, have been identified [
33]. These inflammasomes can be triggered by different stimuli. For instance, the mouse NLRP1b and rat NLRP1 inflammasome sensors can be activated after their cleavage by a lethal factor released by the Gram-positive bacterium
Bacillus anthracis [
34,
35]. NLRP3 mainly recognizes viral dsRNAs, bacterial toxins, reactive oxygen species (ROS) and endogenous damage signals [
32]. NLRC4 responds to bacterial protein stimulation, while AIM2 is predominantly responsible for the recognition of cytoplasmic dsDNAs during bacterial or viral infection [
36,
37]. Pyrin is activated by bacterial toxins that modify RhoA GTPases [
38]. The adaptor protein ASC bridges the interaction between the sensor protein and procaspase-1 within the canonical inflammasome [
39]. ASC recruits procaspase-1 via a CARD–CARD domain interaction [
40]. Remarkably, ASC is indispensable for the pyrin domain-containing sensors (NLRP3, AIM2 and pyrin) to recruit procaspase-1, while the CARD-based sensors (NLRP1b and NLRC4) can directly bind to procaspase-1 [
32]. After being recruited to the inflammasome, procaspase-1 forms dimers and activates its own protease capability to generate caspase-1 [
15]. Caspase-1-mediated cell death represents the canonical pyroptosis pathway. Activated caspase-1 induces the proteolytic processing of the pro-inflammatory precursor cytokines (pro-IL-1β and pro-IL-18) to release active IL-1β and IL-18 [
41]. The pro-pyroptotic factor GSDMD consists of an N-terminal pore-forming domain and a C-terminal repressor domain (RD). The RD domain binds the GSDMD-NT and maintains the protein in an autoinhibitory state [
42]. Caspase-1 activated by the canonical inflammasomes induces the cleavage of GSDMD, liberating the N-terminal fragment (GSDMD-NT) [
11]. In the canonical pyroptosis pathway, the formation of inflammasomes is required for caspase-1-mediated cleavage of GSDMD. Caspase-1, -4, -5 and -11 cleave GSDMD at an aspartate residue in the linker that connects GSDMD-NT and RD, which leads to the generation of a noncovalent GSDMD-NT-RD complex [
43]. Intriguingly, GSDMD-NT has high affinity for specific lipid compositions, such as phosphatidic acid, phosphatidylserine, cardiolipin, mono- and bisphosphorylated phosphoinositols [
44]. As phosphatidylserine and phosphoinositols are restricted to the inner leaflet of the plasma membrane, GSDMD-NT can only oligomerize to form pores from the cytosolic face [
45]. Upon lipid binding, the N-terminal domain of gasdermin A3 (GSDMA3) underwent significant conformational changes, leading to its separation from the RD domain and oligomerization into a ring-shaped structure [
46]. In addition, the conformational changes also facilitated membrane insertion of the ring architecture. Considering the similar structural and biochemical features between GSDMD and GSDMA3, this mechanism could apply to the formation of GSDMD-NT pores. Moreover, cleaved GSDMD exhibits no affinity for the outer leaflet of the cellular membrane, avoiding damage to surrounding cells during pyroptotic cell death [
44]. GSDMD-NT-formed pores mediate osmotic cell swelling, plasma membrane rupture and the liberation of intracellular components including IL-1β and IL-18 [
47]. Additionally, caspase-1 plays an important role in triggering DNA fragmentation.
GSDMD-NT pores act as the conduit for potassium (K
+) efflux that sufficiently triggers the activation of the NLRP3 inflammasome [
48,
49]. Caspase-11 could activate the canonical NLRP3 inflammasome by boosting GSDMD-induced K
+ efflux, demonstrating that canonical and non-canonical inflammasomes functioned synergistically to protect the host against pathogen invasion [
50]. The influx of calcium (Ca
2+) ions from the extracellular environment also occurs through GSDMD-NT-induced pores [
6]. Interestingly, GSDMD-NT pores did not necessarily lead to cell death, since Ca
2+ influx served as a signal for cells to initiate membrane repair program. Moreover, the repair mechanism involved recruitment of the endosomal sorting complexes required for transport (ESCRT) machinery to damaged membrane sites. Accordingly, suppression of the ESCRT-III machinery significantly promoted pyroptotic cell death downstream of GSDMD activation. In the pyroptosis pathway, the GSDMD-NT pore serves as a channel for release of IL-1β and IL-18. Notably, these inflammatory cytokines can be released by alternative mechanisms. For instance, activated caspase-1, pro-IL-1β and pro-IL-18 can be encapsulated into secretory lysosomes [
51]. Caspase-1 processes pro-IL-1β and pro-IL-18 to generate bioactive cytokines within secretory lysosomes. The mature cytokines are then released into the extracellular milieu via fusion of lysosomes with the plasma membrane. Moreover, caspase-1-mediated IL-1β cleavage triggered its translocation from the cytosol to plasma membrane and was sufficient for GSDMD-independent IL-1β release [
52]. In contrast, caspase-1 and GSDMD could accelerate IL-1β secretion. During necroptosis, MLKL activation induced the assembly of the NLRP3 inflammasome and caused plasma membrane rupture [
53]. These events resulted in the maturation and release of IL-1β. Thus, IL-1β and IL-18 can be released into the extracellular space through GSDMD-independent mechanisms.
4. The Emerging Role of Inflammasome-Dependent Cytokines in Cancer Pathogenesis
IL-1β and IL-18 are stored as inactive proforms that reside in the cytoplasm of naive immune cells [
138]. Caspase-1-mediated cleavage of pro-IL-1β and pro-IL-18 occurs during the activation of the inflammasome pathways [
139]. However, aberrantly expressed IL-1β and IL-18 contribute to cancer pathology [
140]. These two cytokines have emerged as pivotal regulators of tumorigenic processes that may either inhibit or promote tumor occurrence, growth, invasion and metastasis according to the tumor stage, type and microenvironment.
4.1. IL-1β and Cancer
The IL-1 signaling cascade is activated upon the binding of IL-1α or IL-1β to the IL-1 receptor type 1 (IL-1R1), recruiting the IL-1R accessory protein (IL-1RAcP) and the myeloid differentiation primary response protein 88 (MyD88) to the receptor complex [
141]. This is followed by the phosphorylation of various kinases and the translocation of NF-ĸB to the nucleus, eventually leading to the activation of inflammatory responses [
142]. IL-1 is a key mediator of innate and adaptive immune responses and plays a critical role in sensing microbial invasion and activating lymphoid cell function [
143]. More importantly, the pro-inflammatory cytokine IL-1β has significant effects on tumor growth, invasiveness and metastasis. For instance, IL-1β was found to be a master cytokine in the development of breast cancer [
144]. Blockage of IL-1β could induce antitumor immunity and resulted in breast cancer regression by activating CD8
+ lymphocytes. OSCC-derived IL-1β favored stromal glycolysis and induced a lactate shuttle to cancer cells, which facilitated the proliferation of OSCC cells [
145]. IL-1β enhanced the proliferation of osteosarcoma (OS) cells by modifying the NF-ĸB/miR-506/Jagged1 (JAG1) pathway [
146]. Reportedly, IL-1β promoted OS cell growth through modulation of the miR-376c/transforming growth factor-α (TGFA) axis [
147]. These studies implied that multiple signaling pathways mediated the pro-tumorigenic activity of IL-1β in OS cells.
IL-1β can adjust the malignant characteristics of cancer cells. IL-1β enhanced the stem-like properties of GC cells by promoting the nuclear translocation of metastasis-promoting S100 calcium-binding protein A4 (S100A4) [
148]. Consistently, blockage of the IL-1β signaling repressed the EMT process in GC cells by upregulating β-catenin and E-cadherin, and downregulating fibronectin, vimentin, Snail, MMP2 and MMP9 [
149]. IL-1β also favored the EMT program in CRC cells by downregulating E-cadherin and upregulating vimentin [
150]. The roles of IL-1β in the invasion of breast cancer were extensively explored. The IL-1β response driven by breast cancer prevented the differentiation of metastasis-initiating cancer cells (MICs) into highly proliferative E-cadherin-positive progeny [
151]. Conversely, abolishment of the pro-inflammatory response led to metastatic colonization of breast cancer. Another study showed that IL-1β induced the EMT process and promoted the malignancy of breast cancer cells by activating the IL-1β/IL-1R1/β-catenin pathway [
152]. Macrophage-derived IL-1β enhanced the migration of breast cancer cells and their adhesion to lymphatic endothelial cells [
153]. IL-1β enhanced the invasion of breast cancer cells via upregulating MMP-9 by activating focal adhesion kinase (FAK) and proto-oncogene tyrosine-protein kinase Src [
154]. IL-1β could enhance the invasion and migration of esophageal squamous cell carcinoma (ESCC) cells by promoting EMT and inducing the NF-ĸB signaling [
155]. IL-1β promoted EMT and metastasis of HCC cells by upregulating hypoxia inducible factor-1α (HIF-1α) [
156]. Similarly, IL-1β induced the nuclear import of NF-ĸB as well as enhanced MMP transcription, thus promoting OSCC invasion and progression [
157]. IL-1β increased the expression of fascin and promoted extracellular matrix degradation and infiltration into the collagen matrix, hence facilitating OSCC cell invasion [
158]. Additionally, IL-1β could increase the expression of glutaredoxin 1 (Grx1) in OSCC cells, thus facilitating the malignant transformation process [
159].
The inflammatory environment is essential for the induction of chemoresistance in cancer cells. The molecular mechanisms underlying the role of the IL-1β signaling pathway in cancer chemoresistance have been disclosed. It was found that IL-1β was able to upregulate the chemoresistance-associated gene, the tumor protein 63 (TP63) isoform ∆NP63α, contributing to the acquisition of cisplatin resistance in breast cancer cells [
160]. IL-1β conferred doxorubicin resistance to breast cancer cells by elevating the expression of the baculoviral inhibitor of apoptosis repeat-containing 3 (BIRC3), known as an EMT marker [
161]. Thus, IL-1β-induced chemoresistance in cancer cells might be attributed to its regulation of the EMT program. IL-1β was able to enhance tamoxifen resistance in breast cancer cells by downregulating the estrogen receptor α (ERα) [
152]. Abrogation of IL-1β enabled PDAC cells to regain gemcitabine sensitivity by targeting IL-1R-associated kinase 4 (IRAK4) [
162]. Altogether, these studies raise the possibility that the IL-1β signaling could be therapeutically disrupted to improve chemotherapeutic efficacy in cancer patients.
4.2. Therapeutic Potential of IL-1 Neutralization in Cancer
IL-1β is considered as an attractive target in cancer treatment. Three IL-1 blockers, including anakinra, canakinumab and rilonacept, have been approved [
163]. The IL-1R antagonist (IL-1Ra) is a natural inhibitor of IL-1β in vivo, where it acts via occupying the IL-1R [
164]. Anakinra is molecularly identical to native IL-1Ra [
165]. IL-1Ra was able to block the IL-1 signaling in chronic myelogenous leukemia (CML) and inhibited the growth of leukemia stem cells (LSCs) [
166]. This study provided a solid basis for further investigation of anti-IL-1 strategies to promote LSC elimination in CML. A randomized phase III trial indicated that the IL-1Ra levels had a significant association with bermekimab responsiveness in patients with advanced CRC [
167]. Anakinra overcame erlotinib resistance in HNSCC xenografts but had no effect on the anticancer activity of erlotinib in HNSCC cells [
168]. The effectiveness of 5-FU plus bevacizumab and anakinra was assessed in patients with metastatic colorectal cancer (mCRC) [
169]. This therapeutic regimen showed good tolerance in mCRC and exhibited efficacy with long-lasting tumor stabilization. Moreover, this combination had a manageable safety profile and might be a promising treatment option for CRC patients. Anakinra plus gemcitabine attenuated the proliferation, invasion and migration of PDAC cells [
170]. Combined treatment with anakinra and gemcitabine also obviously reduced the tumor burden in vivo. Therefore, anakinra in combination with gemcitabine might be an effective therapeutic approach for PDAC.
Canakinumab is a human monoclonal antibody targeting IL-1β [
171]. Reportedly, anakinra or canakinumab repressed breast cancer cell metastasis, and also blocked cancer cells shed into the circulation in vivo [
172]. Anakinra plus canakinumab completely controlled disease activity and inhibited neoplastic recurrence in the patient with refractory Behcet disease uveitis and concomitant bladder papillary carcinoma [
173]. The therapeutic potential of canakinumab in lung cancer patients was previously investigated. A significant decline in the incidence of lung cancer was observed in patients that received 150 or 300 mg of canakinumab as compared to the placebo group [
174]. Lung cancer mortality was remarkably less frequent in patients assigned to canakinumab than that in patients who received the placebo, with the effect being more prominent in the higher dose (300 mg) group [
175]. Rilonacept is a soluble decoy receptor that mainly neutralizes IL-1β [
176]. As expected, rilonacept could maintain inflammatory remission in patients enrolled in a clinical trial [
177]. Specially, rilonacept treatment caused a rapid and sustained decrease in the severity of inflammatory syndromes [
178]. Further clinical studies are demanded to assess the therapeutic effectiveness of rilonacept in cancer.
4.3. IL-18 and Cancer
IL-18 belongs to the IL-1 cytokine family and is constitutively expressed by most cell types including epithelial cells, fibroblasts, macrophages and natural killer (NK) cells [
179]. Like IL-1β, IL-18 is activated when cleaved by caspase-1 following inflammasome activation [
180]. The pro-inflammatory activity of IL-18 is tuned by its physiological inhibitor IL-18 binding protein (IL-18BP) [
181]. IL-18 is involved in the carcinogenesis of multiple cancers. Accumulating evidence indicated that the IL-18 promoter genotype was correlated with the risk of PC, nasopharyngeal carcinoma (NPC), HCC and NSCLC [
182,
183,
184,
185]. Moreover, genetic polymorphisms of IL-18 were linked with the prognosis and survival of patients with acute myeloid leukemia (AML) [
186]. Upregulation of IL-18 was correlated with poor overall survival in patients with multiple myeloma (MM) [
187]. The mechanisms by which this pro-inflammatory cytokine controls cancer progression have been explored. IL-18 restrained the apoptosis of AML cells by elevating the expression of cyclooxygenase-2 (COX-2) [
188]. IL-18 mediated estrogen-related receptor α (ERRα)-regulated proliferation and migration of CRC cells [
189]. IL-18 promoted OSCC cell invasion and metastasis by reinforcing the EMT process via the Wnt/β-catenin signaling pathway [
190]. These studies demonstrated that IL-18 functioned to modify the malignant behaviors of cancer cells. In addition, serum IL-18 levels were evidently lower in patients with pancreatic adenocarcinoma (PA) who had response to gemcitabine-based chemotherapy compared with chemotherapy-unresponsive patients [
191]. Serum IL-18 levels might be used to predict the response to gemcitabine-based chemotherapy in PA patients.
It is well-known that IL-18 acts as a critical participant in initiating antitumor immune responses [
192]. IL-18 modulates innate and adaptive immune responses through the recruitment or differentiation of immune cells, such as NK cells, T cells and monocytes [
179]. Moreover, IL-18 augments IFN-γ production and cytotoxicity of NK cells, T cells and neutrophils [
193]. The previous study demonstrated that IL-18 favored the IFN-γ production by CD8
+ T cells and NK cells, thereby eliciting antitumor immunity during ESCC carcinogenesis [
194]. Accordingly, IL-18 significantly depressed the proliferation and metastasis of ESCC cells [
195]. Likewise, IL-18 facilitated the expansion of NK cells and altered their phenotypes in lung cancer [
196]. IL-18-induced NK cells might be useful for cancer immunotherapy. Adoptive transfer of T cells engineered with a melanoma-specific T cell receptor (TCR) and inducible IL-18 resulted in enhanced antitumor T cell responses, hence suppressing melanoma growth in vivo [
197]. Thus, IL-18 improved the anticancer activity of dacarbazine in malignant melanoma [
198]. IL-18 served a suppressive role in HCC progression by enhancing the differentiation, activity and survival of tumor-infiltrating T cells [
199]. Furthermore, IL-18 inhibited HCC growth by priming NK cells trafficked to the liver [
200]. Mesenchymal stem cells-expressing IL-18 inhibited the proliferation and metastasis of breast cancer cells by activating immunocytes and immune cytokines, downregulating the proliferation marker Ki-67 and suppressing tumor angiogenesis [
201]. Collectively, IL-18-based immunotherapy might be a promising therapeutic strategy for cancer.
On the contrary, IL-18 can compromise host immune responses in favor of cancer evasion. PC cell-derived IL-18 boosted the differentiation of naive B cells into regulatory B cells (Bregs) and ascended expression of programmed cell death-ligand 1 (PD-L1) in Bregs, which led to PC immune tolerance [
202]. Paradoxically, IL-18 enhanced the cytotoxic activity of NK cells and T cells in PC-transplanted mice [
203]. However, IL-18 enhanced the proliferation and invasion of PC cells through the NF-ĸB signaling pathway. When combined with the NF-ĸB inhibitor, IL-18 exhibited a therapeutic effect on PC. It seemed that multiple pathways simultaneously participated in IL-18-regulated PC progression. The complex interplay among these pathways might determine the final effects of IL-18 on cancer pathogenesis. Breast cancer-derived IL-18 triggered programmed cell death-1 (PD-1) expression on immunosuppressive NK cells and was associated with poor prognosis in patients with triple-negative breast cancer [
204]. As expected, IL-18 was implicated in leptin-enhanced breast cancer cell invasion and migration [
205]. Thus, IL-18 could drive breast cancer progression by inducing PD-1-dependent immunosuppression.
4.4. Potential Efficacy of Anti-IL-18 in Cancer Therapy
IL-18 shows anticancer activity in different pre-clinical models of cancer immunotherapy through the activation of NK and/or T cell responses [
206]. Clinical studies have been conducted to assess the therapeutic efficacy of IL-18 in cancer patients. Ten melanoma patients and nine RCC patients were enrolled in a previous study and were assigned to different doses (100, 500, 1000 or 2000 μg/kg) of IL-18 [
207]. No dose-limiting toxicity was observed. Notably, IL-18 exhibited immune regulatory activity in these patients. In a phase I clinical trial, twenty-one RCC patients, six melanoma patients and one patient with Hodgkin lymphoma were given IL-18 in doses ranging from 3 to 1000 μg/kg [
208]. Only one patient administrated with 100 μg/kg IL-18 experienced transient hypotension and bradycardia during the first infusion. No other dose-limiting toxicity was observed. IL-18 administration could effectively activate immune cells (lymphocytes and monocytes). Moreover, the serum levels of IFN-γ, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-18BP and soluble Fas ligand (FasL) were elevated in these patients. Thus, IL-18 had biological effects on the immune system and might be effective in treating cancer. In a phase 2 randomized study, IL-18 was well tolerated and exhibited low toxicity in 64 patients with metastatic melanoma [
209].
However, IL-18 had limited therapeutic efficacy as a single agent in cancer patients. Previously, nineteen patients with non-Hodgkin lymphoma were given rituximab in combination with IL-18 at doses of 1, 3, 10, 20, 30 and 100 μg/kg [
210]. This combination elevated the expression of IFN-γ, GM-CSF, and chemokines. Moreover, objective tumor responses could be observed in five patients. IL-18 (3 μg/kg) plus pegylated liposomal doxorubicin (PLD) was safe and biologically active in sixteen patients with recurrent ovarian cancer [
211]. IL-18 may be used as an immune-stimulatory molecule in combined therapy with conventional chemotherapeutic agents. Intriguingly, IL-18 plays a pro-carcinogenic role in several types of cancer. Under the condition that IL-18 is harmful, the utilization of IL-18BP to neutralize IL-18 may be a potential therapeutic approach for certain types of cancer. A previous study showed that the IL-18BP-Fc therapy restrained the lung metastasis of breast cancer cells by blocking tumor-released IL-18 [
212]. IL-18BP may be an alternative treatment option for cancer patients. These promising results may propel further investigation of the clinical utility of IL-18-based therapy in cancer intervention.
This entry is adapted from the peer-reviewed paper 10.3390/cancers11091313