 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Urszula Demkow | + 3496 word(s) | 3496 | 2021-09-18 17:25:06 | | | |
| 2 | Conner Chen | Meta information modification | 3496 | 2021-09-24 10:54:29 | | |
Video Upload Options
Neutrophils constitute the first line of defense against foreign invaders using major effector mechanisms: phagocytosis, degranulation, and neutrophil extracellular traps (NETs) formation. NETs are composed from decondensed nuclear or mitochondrial DNA decorated with proteases and various inflammatory mediators. Cancer cells recruit neutrophils (tumor-associated neutrophils, TANs), releasing NETs to the tumor microenvironment. NETs were found in various samples of human and animal tumors. The role of the NETs in tumor development increasingly includes cancer immunoediting and interactions between the immune system and cancer cells. According to the accumulated evidence, NETs awake dormant cancer cells, causing tumor relapse, as well as its unconstrained growth and spread. NETs play a key regulatory role in the tumor microenvironment, such as the development of distant metastases through the secretion of proteases, i.e., matrix metalloproteinases and proinflammatory cytokines. NETs, furthermore, directly exacerbate tumor aggressiveness by enhancing cancer migration and invasion capacity.
1. Neutrophils and NETs
Polymorphonuclear neutrophils (PMNs), the most abundant white blood cells, are frontline fighters against invading microorganisms. PMNs destroys pathogens, or other endogenous or exogenous factors, using a combination of mechanisms, including phagocytosis, oxidative bursts, the release of antimicrobial mediators, and the production of neutrophil extracellular traps (NETs) [1]. NETs are web-like structures built from nuclear or mitochondrial DNA fibers, decorated with anti-microbial enzymes and histones, which are released to entrap and kill pathogens [2]. Besides their role as an anti-microbial weapon, NETs create a physical barrier for both pathogens and immune cells. The process of NET formation in its classical form is called NETosis and has been defined as a type of regulated cell death distinguished from apoptosis and necrosis [3]. Further studies have described an alternative pathway of a non-cell-death NETs generation, named vital NETosis. NET release is initiated by an oxidative burst via raf-MEK-ERK activation of NADPH oxidase. Subsequently, neutrophil elastase (NE) translocates from azurophil granules into the nucleus, where it instigates chromatin breakdown through histone hydrolysis. Further observations have suggested that myeloperoxidase (MPO) has also been implicated in chromatin decondensation and the rupturing of the nuclear envelope. Chromatin decompaction is further supported by peptidyl arginine deiminase 4 (PAD4)—a protein-citrullinating enzyme that enters the nucleus to deiminate specific arginine residues on histones, resulting in the loss of positive charge from the transformed arginine residues and the disassembling of nucleosome structure [4]. Crucial steps in NET formation include nuclear swelling, nuclear envelope disintegration, the mixing of nucleic acids and granule proteins within a large intracellular vacuole, the spilling of nuclear content into the cytoplasm, and, finally, cell membrane breakdown [5].
2. NETs Fuel Cancer Progression and Indicate Poor Prognosis
2.1. How Do NETs Awaken Dormant Cancer Cells?
Cancer cells from a primary tumor can migrate to other tissues, remaining dormant and clinically silent for a long time. The concept of tumor cell dormancy has been described for most common solid cancers, including breast, prostate, lung, colon, and kidney cancers, as well as melanoma. Hematological malignancies, such as multiple myeloma, lymphoma, and leukemia, were included as well [6]. The slow-cycling cancer cells can disseminate early and seed secondary organs where they wait to be awakened, thus causing cancer to recur. Dormant cancer cells settle in specific niches. For example, breast cancer cells inhabit the perivascular regions of the lung [6]. The exact mechanisms causing the awakening, the restart of proliferation, and the metastasis of the slow-cycling cells overlooked by the immune system (immune evasion) are largely unknown. It has been reported that NETs possess the ability to wake dormant cancer cells, and are thus responsible for tumor relapse and metastatic spread [7]. Consistently, NETs formed in the course of the inflammatory process have awakened malignant cells in experimental tumor models. In an excellent study Albrengues et al. proved that NETs released in the course of chronic pulmonary inflammation awaken dormant breast cancer cells and promote metastatic spread [7]. The chronic lung inflammation in this model was induced by infection or cigarette smoke. Using a cell cycle reporter to measure dormancy against the reactivation of cancer cells, these authors found that prolonged inflammation induced by repeated lipopolysaccharide (LPS) inhalation caused dormant cancer cells to restart proliferation, and this process was dependent on the presence of intact neutrophils. The dormant malignant cells could be awakened by LPS even a month after they had inhabited the lungs. The effect of NETs on the cancer was exerted indirectly via extracellular matrix (ECM) remodeling. The analysis revealed that NET proteinases, NE and MMP9, cleaved laminin, revealing new epitopes of this molecule. Such modified laminin activated integrin α3β1, which in turn re-initiated cancer cell proliferation. The researchers confirmed the presence of cycling cells close to remodeled laminin, and on the contrary, the cells near intact laminin remained dormant. Blocking the new epitope of laminin with dedicated antibodies hindered the awakening of cancer cells, both in vitro and in vivo. Furthermore PAD 4 inhibitor or DNAase treatment impeded the formation of NETs and prevented the activation of quiescent cells and metastasis formation [7]. Recent discoveries have suggested that NE and MMP9 blockades in vitro prevent cancer from re-entering cell cycle and block LPS-mediated cancer progression in vivo. Furthermore, inhibiting NET formation further prevented neutrophil accumulation, thus breaking the vicious cycle of self-perpetuating inflammation. These effects were also reported by studies of Orgaz et al., who revealed that NET proteases, such as MMP9, are associated with metastatic dissemination [8]. Continuing this experimental work, Albrengues et al. found that not only laminin, but also thrombospondin-1 (TSP-1), was disintegrated by NE and MMP9 [7]. TSP-1 upregulates integrin 6 subunit expression, thus promoting tumor cell adhesion to laminin, and subsequently supporting malignant cell invasion [9]. The observations of Albrengues et al. suggest that TSP-1 abolished the effect exerted by cleaved laminin-111 on cell proliferation, thus, TSP-1 prevented metastatic relapse by proteolytic remodelling of laminin-111 [7]. Albrengues et al. thus concluded that both TSP-1 degradation and laminin remodeling are necessary to awake quiescent cells in their niches. Integrin β1 accounts for the activation of FAK-ERK-MLC2-YAP signaling pathway, contributing to proliferation and survival of malignant cells. In accord with this observation, NET-induced activation of the same pathway, requiring NE and MMP9 activity, awakes slow-cycling cancer cells. Whatever the precise mechanistic basis of this process may be, experiments with RNAi silencing suggest that α3β1 integrin and transcriptional regulator YAP in cancer cells are necessary for NET-dependent awakening of dormant cancer cells. The study of Albrengues et al. confirmed the hypothesis of “seed and soil”, i.e., the predilection for metastasis to specific organs where the local microenvironment is favorable [7][10]. Amongst the many components of the tumor microenvironment (soil), neutrophils, and their products, all play a prominent role in tumor (seeds) progression, the evasion of the immune system, and metastasis [7].
2.2. How NETs Promote Cancer Invasion, Evasion, Its Spread, and Metastasis Formation
The systemic spread and formation of metastases in distant organs is responsible for the majority of cancer deaths. A multi-step process of metastasis formation includes local invasion, intravasation, and the survival of tumor cells in the circulation, which is followed by extravasation from blood or lymphatic vessels, the colonization of distant sites, the awakening from dormancy, and the metastatic spread. At each step of this complex process, malignant cells must also resist attacks from the host’s immune system. There is experimental evidence suggesting that NETs participate at every stage of this process, given their versatile role in the metastatic cascade. (Figure 1).
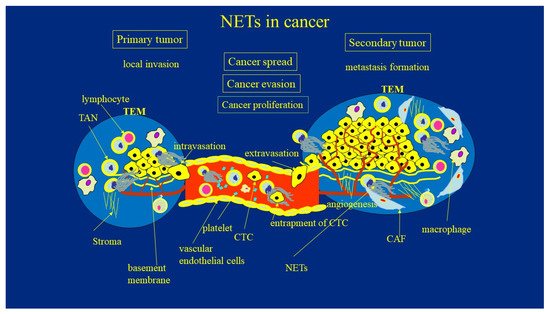
Figure 1. The role of NETs in cancer development. TAN—tumor associate neutrophils; TEM—tumor environment; CAF—cancer associated fibroblasts; NETs—neutrophil extracellular traps; CTC—circulating tumor cells.
2.3. NETs Supports the Cancer Evasion Strategies
The evasion of tumor cells from immunosurveillance depends on the interplay between various infiltrating immune cells and tumor cells. Whatever the mechanistic basis, it appears that immunosurveillance of tumors is canonically dependent on the presence of the major histocompatibility complex 1 (MHC1) antigens on cancer cells enabling lymphocytes T (both CD4+ and CD8+) to discriminate tumor cells from normal cells, as well as to control the tumor cell survival [11][12]. Various proteolytic enzymes (proteinases) are able to modulate the cell-surface-associated presentation of MHC molecules. MMP9, for example, is responsible for the shedding of MHC class I antigen from cancer cells [13]. The selected subclones of malignant cells achieve the capability to hide from the immune system by losing the ability to present cancer antigens to T-cells. Various components of ECM have been recognized as sources of signals for the immune system to slow down immune reactions, for example through the expression of checkpoint molecules. The ECM is a reservoir of immunomodulatory cytokines and growth factors that are released upon their proteolytic degradation. Metalloproteinases and NE can modulate immune and inflammatory responses through the degradation of the ECM. The cleavage products of the ECM (e.g., matrikines) can, by themselves, affect immune surveillance. NET proteases can impede the immune response and, thus, ensure the best possibility of cancer cell survival by enabling the metastatic process [7]. According to Albrengues et al. [7], the degradation of matrix proteins is one of the mechanisms of tumor evasion that silences the host’s immune system. NET proteinases stimulate the production of IL-8, IL-1β, and TNF-α with tumor-associated macrophages through the activation of several MMPs. This process is dependent on Src kinase activation, highlighting the fact that NE also impacts integrins and integrin-mediated intracellular signaling [14][15]. As another example, the inhibition of hyaluronic acid (a major component of the ECM) synthesis by 4-methylumbelliferone in a mesothelioma xenograft has led to a significant increase in the expression of both immune checkpoint molecules, PD-1 and PD-L1 [16]. Although the mechanisms involved in ECM modification by NET components are not fully elucidated, the clear connection between the ECM composition and proteinases, as well as the immune escape, strongly support the existence of such an effect. Onuma et al. [17] confirmed that the blockade of NETs, in combination with immune checkpoint PD-1 inhibition, improved the response rates of colorectal cancer metastases to immune checkpoint inhibitors as a single therapy. This was achieved through the improving of the function of exhausted CD8+ T-cells [17].
2.4. NETs Enhance Invasion Capacity of Cancer Cells
A crucial event at the first stage of metastatic colonization is the formation of a favorable niche for tumor engraftment attributable to tumor–stroma crosstalk [18]. The process of metastatic spread began from proteolytic remodeling of ECM and the release of ECM metabolites necessary, or even mandatory, for the dissemination of cancer cells. As mentioned above, the ECM is digested by MMPs, disintegrin, metalloproteinases with thrombospondin motifs (ADAMTS), and proteases that specifically cleave at cysteine, serine, and threonine residues [19]. Several components of mature NETs cause an imbalance in the microenvironments, as well as the emergence of metastatic niches. For example, NET-derived NE and MMP-9 degraded ECM to actively induce tumor invasion [20]. Accordingly, it was shown that matrix metalloproteinase catalytic activity modulated the invasiveness and provided a route for the malignant cells to metastasize via modulation of the integrins–FAK signaling pathway [21]. In an experimental model using Boyden transwell invasion assay, Park et al. [22] focused on the neutrophil-mediated invasion of tumor cells. The applied model confirmed that tumor invasion through the filter in the transwell system can be promoted by the mutual interaction between tumor cells in the upper chamber and neutrophils in the lower chamber. Furthermore, the blockade of NE and matrix metalloproteinases impeded tumor invasion [22]. In agreement with this observation, DNase I treatment downregulated NE and NET activities and reduced the invasive and metastatic potential of malignant cells [22]. Other investigators were able to confirm significant correlation between NETs and liver metastases of patients with breast and colon cancers, thus confirming increased binding activity of transmembrane protein CCDC25 on primary cancer cells to NET DNA. These authors proved that CCDC25 senses extracellular DNA and, subsequently, activates the ILK-β-parvin pathway to attract cancer cells. NET-mediated metastasis was abrogated in CCDC25-knockout cells. Moreover, the expression of CCDC25 was associated with a poor outcome of the disease [23]. Although the detailed mechanism of tumor invasion and metastasis via NET molecules is still not completely understood, it would be interesting to investigate the role of TANs in the regulation of NET-mediated tumor invasion. Signaling is an integral process in controlling invasive and metastatic potential of tumor cells. The signaling between various structures in TME, including NETs fragments, is crucial in controlling the invasive potential of the tumor. Thus, in silico studies modelling these critical interactions and their effects are warranted to discern alternative explanations of these processes and pave the way for the development of new therapeutic strategies [24].
2.5. NETs Enhance Systemic Spread and Tumor-Associated Angiogenesis
Tumor cells can migrate and intravasate the blood or lymph vasculature. They can survive within the circulation, then extravasate at distant sites. The factors determining adhesion strength, which might influence the ability of cells to transmigrate through an endothelial cell monolayer and the basement membrane, are poorly understood. Recent studies have highlighted that these processes are driven not only by signals from cancer cells, but are also modified by signals from components of the TME [18]. Current evidence suggests that NETs may play a crucial role in the hematogenous spread of tumors. Jung et al. [25] showed that NETs promoted tumor growth, metastasis, and angiogenesis of the pancreatic cancer cell line (AsPC-1). NETs used as chemoattractans stimulated AsPC-1 cell migration (in a Matrigel-coated invasion chamber) better then intact neutrophils. These effects were abrogated by histone-binding agents (heparin, polysialic acid), DNAse I, and Toll-like receptor neutralizing antibodies. Antibodies against both TLR2 and TLR4 significantly inhibited NET-mediated AsPC-1 cell migration. Although not unexpectedly, these results support the opinion that TLR2 and TLR4 participate in tumor transmigration. In patients with pancreatobiliary malignancy, elevated NET markers correlated with hypercoagulability makers. Histone–DNA complexes were used as markers of NETs. Another component of NETs, histones, significantly increased the endothelial cell proliferation and the formation of new blood vessels in a dose-dependent manner. Application of histone-binding agents abrogated histone-induced angiogenesis [25]. The same directionality of the effect was observed by Tohme et al., who reported that the chemotactic factor released during NET formation may stimulate proliferation and migration of cancer cells [26]. Finally, the transmigration mechanisms were explained by Kołaczkowska et al., who observed the adherence of circulating NETs to blood, resulting in increased cancer extravasation efficiency, which would enable cancer cells to cross the endothelial barrier [27]. On the other hand, the previously mentioned report of Park et al. suggested that not DNA itself, but rather NET-related proteases are responsible for this effect [22]. Such a discrepancy may be explained by the fact that such a structure as complex as the one between NETs and the locally concentrated enzymes, must be taken as an inseparable assembly, rather than a conglomerate of individual components. Once in the circulation, tumor cells become entrapped by NETs DNA threads. Through the use of cecal ligation, Cools-Lartigue et al. [28] demonstrated the presence of circulating lung carcinoma cells wrapped in NET DNA conglomerates in a murine model of infection. Consequently, circulating “packages” were seeded in the liver, forming micrometastases within 48 h and secondary liver cancer 2 weeks after the cancer cell injection. DNAse or NE inhibitors abrogated the effects [28]. Evidence consistent with these observations was provided by Najmeh et al. from the same group, who found a significant association between upregulation of β1-integrin and NET-related entrapment of circulating lung carcinoma cells, further facilitating metastasis formation and cancer spread [29]. Whatever the precise basis of this mechanism is, it appears that inflammatory mediators harbored by neutrophils may be responsible for insufficient clearance of circulating cells [30]. NETs’ entrapping abilities can be, at least partially, attributed to the ability to adhere to DNA mesh carried by the variety of integrins expressed on the surface of cancer cells. Such interaction was completely abrogated by DNase 1 [31]. Furthermore, the TAN-CTC adhesion process facilitates cancer cell extravasation through the breaking of the transendothelial barrier [32]. The proposed adhesive interaction between circulating neoplastic cells and TANs leads to the increased endothelial cell contraction, permeability, and malignant cell extravasation [32]. A multi-level model shed new light on the fundamental processes elucidating the role of NETs in cancer invasions, transport, and transendothelial migration, thus taking into account specific NET–cell adhesion, ECM–tumor–NET interaction, and intracellular signaling [33][34][35][36][37]. Further studies, however, are still warranted to explore these issues.
2.6. How NETs and Tumor Communicate
The interaction between the tumor and NETs is reciprocal. In their excellent paper, Demeters et al. compiled initial reports showing that TANs are a potent source of NETs and, on the other hand, cancer cells can stimulate neutrophils to release NETs as shown in various animal models of cancer [38]. NETs enhance the gathering and proliferation of single cancer cells, contributing to tumor metastasis by releasing MMP and NE, which through the degradation of ECM, paves a way for tumor cells to leave the primary niche and to migrate to other organs. Conversely, inflammatory cytokines, such as IL-8 and granulocyte colony-stimulating factor, as well as various soluble factors, i.e., exosomes released from cancer cells, stimulate neutrophils to release NETs [39]. Metastatic cancer cells possess the ability to stimulate the release of NETs directly and without the engagement of inflammatory mechanisms [40]. According to the model of a vicious circle proposed by Park et al., the metastatic breast cancer cells induced neutrophils to form NETs, which further enhanced tumor cell growth in target organs [22]. McInturff et al. demonstrated that cancer cells themselves are able to stimulate neutrophils to form NETs in a hypoxic environment where solid tumor growth is enhanced by the higher expression of HIF-1α [41]. Another mechanism by which cancer cells may stimulate neutrophils to form NETs depends on the production of IL-8 and the release of exosomes which require additional priming with granulocyte colony-stimulating factors. Leal et al. found that tumor-derived exosomes of cancer patients in a hypercoagulable state can induce NET release, and that NETs can serve as a scaffold for coagulation factors, platelets, and exosomes carrying prothrombotic mediators, altogether promoting the development of thrombo-embolic complications and cancer progression [39]. In an excellent review, Yousefi et al. summarized various experimental evidence that lung, colon, ovarian, and anaplastic thyroid cancer (ATC) cells induce the release of mitochondrial extracellular DNA traps by viable neutrophils [42]. Furthermore, tumor cells have been demonstrated to produce IL-8, attracting myeloid-derived suppressor cells and activating neutrophil precursors to release NETs [43]. Similarly, liver ischemia reperfusion in a murine model resulted in NET extrusion in parallel with the progression of metastatic disease, while the pre-treatment of mice with topical DNase or a PAD4 inhibitor abrogated these effects [26]. Consistently with these observations in mice, an increased postoperative NET formation inversely correlated with the disease-free survival in patients undergoing liver resection for metastatic colorectal cancer [26]. However, the limitation of this study manifested in the use of NET plasma markers (MPO–DNA complexes) as surrogates of netting capacities of neutrophils rather than a direct analysis of NET presence in the examined tissues.
2.7. NETs in the Formation of Metastatic Niche
Tumors metastasize to distant organs with tissue-specific microenvironments, which are very different from that of a primary tumor. The precondition of distinct microenvironments involving ECM remodeling and the creation of a favorable pre-metastatic niche is necessary for the seeding of new tumor colonies [19]. The most common modification of the ECM in the primary TME is increased collagen deposition. On the contrary, fibronectin dominates along with glycoproteins and proteoglycans such as tenascin C, osteopontin, and versican in a pre-metastatic niche [44]. The primary niche is mainly formatted by mediators released by growing tumor cells, further acting on various components of TME, which in turn release a second generation of molecules, directly creating a favorable microenvironment. NETs participate in this process, conferring the effect on the electrostatic charge and conformation of fibronectin and collagens in the process of citrullination. This effect is mediated by the enzyme PAD4, derived from NETs during pre-metastatic niche formation [45][46]. Moreover, NETs equipped with proteases are highly associated with aggressive tumor growth and invasion, but this high metastatic potential is abrogated by DNase I treatment [24][22]. Recently, different studies began to highlight that epithelial–mesenchymal transition (EMT), a process by which epithelial cells acquire mesenchymal properties endowing cancer cells with invasive and metastatic potential, is driven by NETs [47][48]. Martins-Cardoso et al. recently described the association between NETs and the pro-metastatic phenotype of human breast cancer cells [47]. Co-cultures of tumor cells treated with isolated NETs underwent several experiments, including migration assay, quantitative RT-PCR, Western blotting, immunofluorescence, and flow cytometry assays. RNA-seq data from The Cancer Genome Atlas (TCGA) database were also assessed [48]. NET components changed the epithelial into mesenchymal phenotype (upregulated expression of N-cadherin and fibronectin, and downregulation of E-cadherin). The effect was accompanied by the increased motility of cells. RNA-seq revealed pro-inflammatory and pro-metastatic signatures. Accordingly, TCGA data analysis of samples from breast cancer patients showed a significant correlation between neutrophil and the pro-tumoral signature of gene expression [47]. Further studies have shed light on the crosstalk between glioma progression and NETs in TME. The tumor growth was mediated via the HMGB1/RAGE/IL-8 axis [49]. Covid-era discoveries also led to the conclusion that lung inflammation and a cytokine storm accompanied by NET formation in the course of COVID-19 contributes to dormant cancer cells awakening and the formation of a pro-metastatic niche [50].
References
- Teng, T.S.; Ji, A.L.; Ji, X.Y.; Li, Y.Z. Neutrophils and Immunity: From Bactericidal Action to Being Conquered. J. Immunol. Res. 2017, 2017, 9671604.
- Pruchniak, M.P.; Demkow, U. Potent NETosis inducers do not show synergistic effects in vitro. Cent. Eur. J. Immunol. 2019, 44, 51–58.
- Manda-Handzlik, A.; Bystrzycka, W.; Cieloch, A.; Glodkowska-Mrowka, E.; Jankowska-Steifer, E.; Heropolitanska-Pliszka, E.; Skrobot, A.; Muchowicz, A.; Ciepiela, O.; Wachowska, M.; et al. Nitric oxide and peroxynitrite trigger and enhance release of neutrophil extracellular traps. Cell Mol. Life Sci. 2020, 77, 3059–3075.
- Manda-Handzlik, A.; Fiok, K.; Cieloch, A.; Heropolitanska-Pliszka, E.; Demkow, U. Convolutional Neural Networks-Based Image Analysis for the Detection and Quantification of Neutrophil Extracellular Traps. Cells 2020, 9, 508.
- Leliefeld, P.H.C.; Koenderman, L.; Pillay, J. How Neutrophils Shape Adaptive Immune Responses. Front. Immunol. 2015, 6, 471.
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411.
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Kuttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227.
- Orgaz, J.; Pandya, P.; Dalmeida, R.; Karagiannis, P.; Sanchez-Laorden, B.; Viros, A.; Albrengues, J.; Nestle, F.O.; Ridley, A.J.; Gaggioli, C.; et al. Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat. Commun. 2014, 5, 4255.
- Sargiannidou, I.; Qiu, C.; Tuszynski, G.P. Mechanisms of thrombospondin-1-mediated metastasis and angiogenesis. Semin. Thromb. Hemost. 2004, 30, 127–136.
- Sanz-Moreno, V.; Balkwill, F.R. Mets and NETs: The Awakening Force. Immunity 2018, 49, 798–800.
- Hoenicke, L.; Zender, L. Immune surveillance of senescent cells–biological significance in cancer- and non-cancer pathologies. Carcinogenesis 2012, 33, 1123–1126.
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111.
- Chitadze, G.; Lettau, M.; Bhat, J.; Wesch, D.; Steinle, A.; Furst, D.; Mytilineos, J.; Kalthoff, H.; Janssen, O.; Oberg, H.H.; et al. Shedding of endogenous MHC class I-related chain molecules A and B from different human tumor entities: Heterogeneous involvement of the a disintegrin and metalloproteases 10 and 17. Int. J. Cancer 2013, 133, 1557–1566.
- Tolle, F.; Umansky, V.; Utikal, J.; Kreis, S.; Bréchard, S. Neutrophils in Tumorigenesis: Missing Targets for Successful Next Generation Cancer Therapies? Int. J. Mol. Sci. 2021, 22, 6744.
- Krotova, K.; Khodayari, N.; Oshins, R.; Aslanidi, G.; Brantly, M.L. Neutrophil elastase promotes macrophage cell adhesion and cytokine production through the integrin-Src kinases pathway. Sci. Rep. 2020, 10, 15874.
- Cho, H.; Matsumoto, S.; Fujita, Y.; Kuroda, A.; Menju, T.; Sonobe, M.; Kondo, N.; Torii, I.; Nakano, T.; Lara, P.N.; et al. Trametinib plus 4-methylumbelliferone exhibits antitumor effects by ERK blockade and CD44 downregulation and affects PD-1 and PD-L1 in malignant pleural mesothelioma. J. Thorac. Oncol. 2017, 12, 477–490.
- Onuma, A.; He, J.; Xia, Y.; Zhang, H.; Genkin, D.; Tetz, G.; Huang, H. and Tsung, A. Neutrophil extracellular traps blockade in combination with PD-1 inhibition in treatment of colorectal cancer metastasis. J. Clin. Oncol. 2020, 38 (Suppl. 15), e16002.
- Leach, J.; Morton, J.P.; Sansom, O.J. Neutrophils: Homing in on the myeloid mechanisms of metastasis. Mol. Immunol. 2019, 110, 69–76.
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120.
- Brostjan, C.; Oehler, R. The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov. 2020, 6, 26.
- Das, A.; Monteiro, M.; Barai, A.; Kumar, S.; Sen, S. MMP Proteolytic Activity Regulates Cancer Invasiveness by Modulating Integrins. Sci. Rep. 2017, 7, 14219.
- Park, J.; Wysocki, R.W.; Amoozgar, Z.; Maiorino, L.; Fein, M.R.; Jorns, J.; Schott, A.F.; Kinugasa-Katayama, Y.; Lee, Y.; Won, N.H.; et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci. Transl. Med. 2016, 8, 361ra138.
- Yang, L.; Liu, Q.; Zhang, X.; Liu, X.; Zhou, B.; Chen, J.; Huang, D.; Li, J.; Li, H.; Chen, F.; et al. DNA of Neutrophil Extracellular Traps Promotes Cancer Metastasis via CCDC25. Nature 2020, 583, 133–138.
- Lee, J.; Lee, D.; Lawler, S.; Kim, Y. Role of neutrophil extracellular traps in regulation of lung cancer invasion and metastasis: Structural insights from a computational model. PLoS Comput. Biol. 2021, 17, e1008257.
- Jung, H.S.; Gu, J.; Kim, J.-E.; Nam, Y.; Song, J.W.; Kim, H.K. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS ONE 2019, 14, e0216055.
- Tohme, S.; Yazdani, H.O.; Al-Khafaji, A.B.; Chidi, A.P.; Loughran, P.; Mowen, K.; Wang, Y.; Simmons, R.L.; Huang, H.; Tsung, A. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016, 76, 1367–1380.
- Kolaczkowska, E.; Jenne, C.N.; Surewaard, B.G.; Thanabalasuriar, A.; Lee, W.Y.; Sanz, M.J.; Mowen, K.; Opdenakker, G.; Kubes, P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat. Commun. 2015, 6, 6673.
- Cools-Lartigue, J.; Spicer, J.; McDonald, B.; Gowing, S.; Chow, S.; Giannias, B.; Bourdeau, F.; Kubes, P.; Ferri, L. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J. Clin. Investig. 2013, 123, 3446–3458.
- Najmeh, S.; Cools-Lartigue, J.; Rayes, R.F.; Gowing, S.; Vourtzoumis, P.; Bourdeau, F.; Giannias, B.; Berube, J.; Rousseau, S.; Ferri, L.E.; et al. Neutrophil extracellular traps sequester circulating tumor cells via β1-integrin mediated interactions. Int. J. Cancer 2017, 140, 2321–2330.
- Roth, S.; Agthe, M.; Eickhoff, S.; Möller, S.; Karsten, C.M.; Borregaard, N.; Solbach, W.; Laskay, T. Secondary necrotic neutrophils release interleukin-16C and macrophage migration inhibitory factor from stores in the cytosol. Cell Death Discov. 2015, 1, 15056.
- Monti, M.; De Rosa, V.; Iommelli, F.; Carriero, M.V.; Terlizzi, C.; Camerlingo, R.; Belli, S.; Fonti, R.; Di Minno, G.; Del Vecchio, S. Neutrophil extracellular traps as an adhesion substrate for different tumor cells expressing RGD-binding integrins. Int. J. Mol. Sci. 2018, 19, 2350.
- Reymond, N.; d’Agua, B.B.; Ridley, A.J. Crossing the endothelial barrier during metastasis. Nat. Rev. Cancer 2013, 13, 858–870.
- Kim, Y.; Stolarska, M.; Othmer, H.G. A hybrid model for tumor spheroid growth in vitro I: Theoretical development and early results. Appl. Sci. 2007, 17, 1773–1798.
- Kim, Y.; Kang, H.; Powathil, G.; Kim, H.; Trucu, D.; Lee, W.; Lawler, S.; Chaplain, M. Role of extracellular matrix and microenvironment in regulation of tumor growth and LAR-mediated invasion in glioblastoma. PLoS ONE 2018, 13, e0204865.
- Kim, Y.; Powathil, G.; Kang, H.; Trucu, D.; Kim, H.; Lawler, S.; Chaplain, M. Strategies of eradicating glioma cells: A multi-scale mathematical model with miR-451-AMPK-mTOR control. PLoS ONE 2015, 10, e0114370.
- Kim, Y.; Othmer, H.G. A hybrid model of tumor-stromal interactions in breast cancer. Bull. Math. Biol. 2013, 75, 1304–1350.
- Lee, W.; Lim, S.; Kim, Y. The role of myosin II in glioma invasion: A mathematical model. PLoS ONE 2017, 12, e0171312.
- Demers, M.; Krause, D.S.; Schatzberg, D.; Martinod, K.; Voorhees, J.R.; Fuchs, T.A.; Scadden, D.T.; Wagner, D.D. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc. Natl. Acad. Sci. USA 2012, 109, 13076–13081.
- Leal, A.C.; Mizurini, D.M.; Gomes, T.; Rochael, N.C.; Saraiva, E.M.; Dias, M.S.; Werneck, C.C.; Sielski, M.S.; Vicente, C.P.; Monteiro, R.Q. Tumor-Derived Exosomes Induce the Formation of Neutrophil Extracellular Traps: Implications for the Establishment of Cancer-Associated Thrombosis. Sci. Rep. 2017, 7, 6438.
- Cedervall, J.; Hamidi, A.; Olsson, A.-K. Platelets, NETs and cancer. Thromb. Res. 2018, 164, 148–152.
- McInturff, A.M.; Cody, M.J.; Elliott, E.A.; Glenn, J.W.; Rowley, J.W.; Rondina, M.T.; Yost, C.C. Mammalian target of rapamycin regulates neutrophil extracellular trap formation via induction of hypoxia-inducible factor 1 alpha. Blood 2012, 120, 3118–3125.
- Yousefi, S.; Simon, D.; Stojkov, D.; Karsonova, A.; Karaulov, A.; Simon, H.U. In vivo evidence for extracellular DNA trap formation. Cell Death Dis. 2020, 11, 300.
- Alfaro, C.; Teijeira, A.; Oñate, C.; Pérez, G.; Sanmamed, M.F.; Andueza, M.P.; Alignani, D.; Labiano, S.; Azpilikueta, A.; Rodriguez-Paulete, A.; et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin. Cancer 2016, 22, 3924–3936.
- Paolillo, M.; Schinelli, S. Extracellular Matrix Alterations in Metastatic Processes. Int. J. Mol. Sci. 2019, 20, 4947.
- Cedervall, J.; Zhang, Y.; Huang, H.; Zhang, L.; Femel, J.; Dimberg, A.; Olsson, A.K. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res. 2015, 75, 2653–2662.
- Masucci, M.T.; Minopoli, M.; Del Vecchio, S.; Carriero, M.V. The Emerging Role of Neutrophil Extracellular Traps (NETs) in Tumor Progression and Metastasis. Front. Immunol. 2020, 11, 1749.
- Martins-Cardoso, K.; Almeida, V.H.; Bagri, K.M.; Rossi, M.I.D.; Mermelstein, C.S.; König, S.; Monteiro, R.Q. Neutrophil Extracellular Traps (NETs) Promote Pro-Metastatic Phenotype in Human Breast Cancer Cells through Epithelial–Mesenchymal Transition. Cancers 2020, 12, 1542.
- Krasnov, G.S.; Kudryavtseva, A.V.; Snezhkina, A.V.; Lakunina, V.A.; Beniaminov, A.D.; Melnikova, N.V.; Dmitriev, A.A. Pan-Cancer Analysis of TCGA Data Revealed Promising Reference Genes for qPCR Normalization. Front. Genet. 2019, 10, 97.
- Zha, C.; Meng, X.; Li, L.; Mi, S.; Qian, D.; Li, Z.; Wu, P.; Hu, S.; Zhao, S.; Cai, J.; et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol. Med. 2020, 17, 154–168.
- Francescangeli, F.; Laura De Angelis, M.; Zeuner, A. COVID-19: A potential driver of immune-mediated breast cancer recurrence? Breast Cancer Res. 2020, 22, 117.

