 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sergiusz Lulinski | + 2148 word(s) | 2148 | 2021-09-14 10:18:51 | | | |
| 2 | Rita Xu | Meta information modification | 2148 | 2021-09-22 04:09:05 | | |
Video Upload Options
Heteroelement analogues of benzoxaboroles constitute an interesting class of boracyclic compounds and may offer the opportunity for various applications while retaining high stability arising from the presence of a strong B-O bond in the ring structure. The replacement of a carbon atom in the boracycle or an adjacent benzene ring with a heteroatom may result in a significant change of structural behaviour. Moreover, physicochemical properties, including solubility, lipophilicity, hydrolytic stability, boron Lewis acidity, and others, can be modified. The aim of this review is to highlight several emerging groups of boracyclic systems which comprise various heteroelement atoms such as another boron, silicon, tin, nitrogen, phosphorus, and iodine. The information on synthesis and properties of such systems is complemented by presentation of their practical potential encompassing especially organic synthesis and catalysis as as medicinal chemistry.
1. Introduction
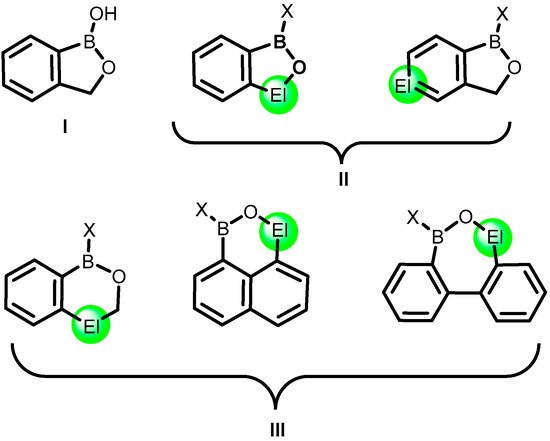
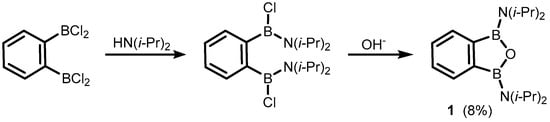
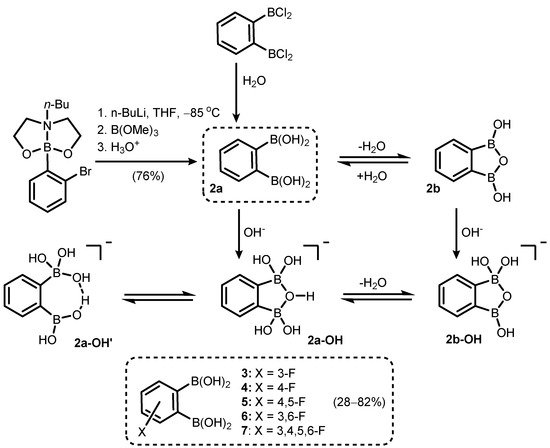
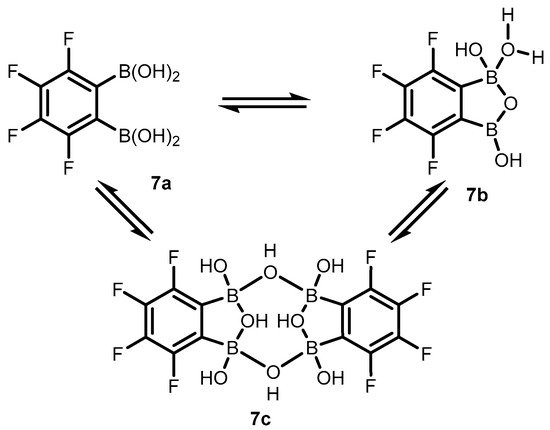
2. Benzoxadiboroles and Related Ring-Expanded Systems Comprising B-O-B Linkage
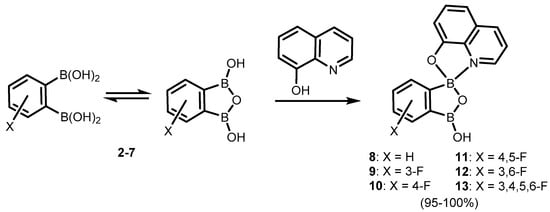
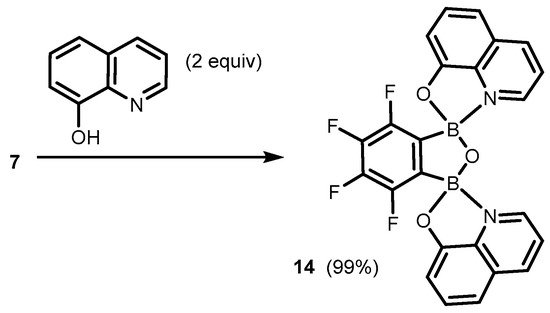
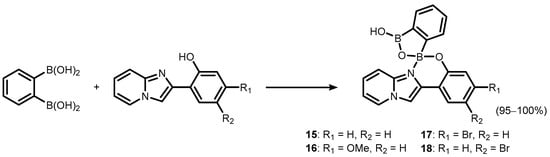
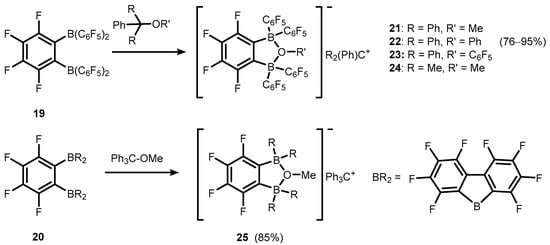
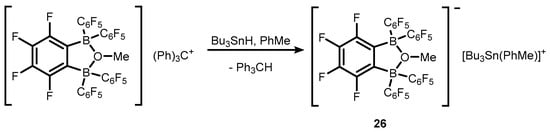
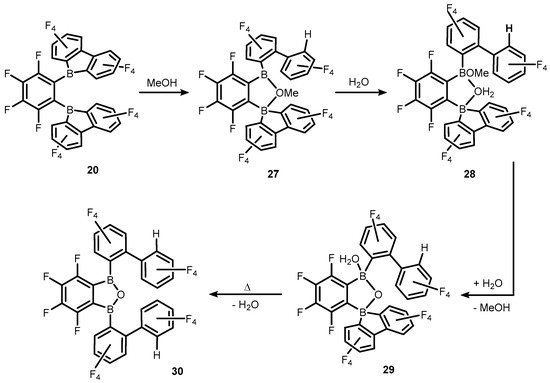
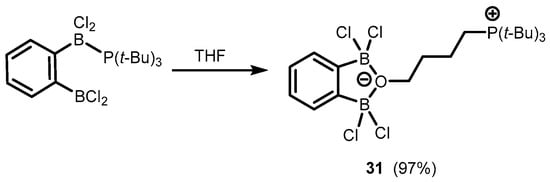
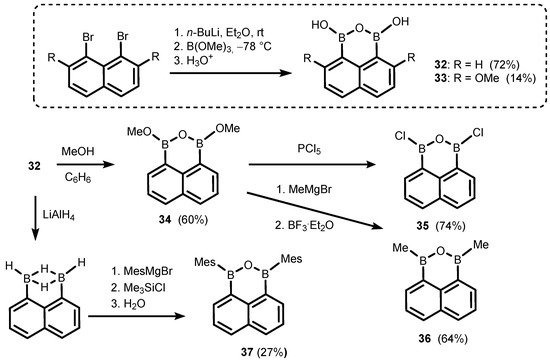
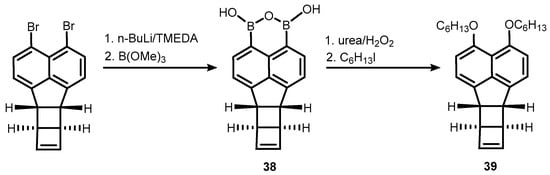
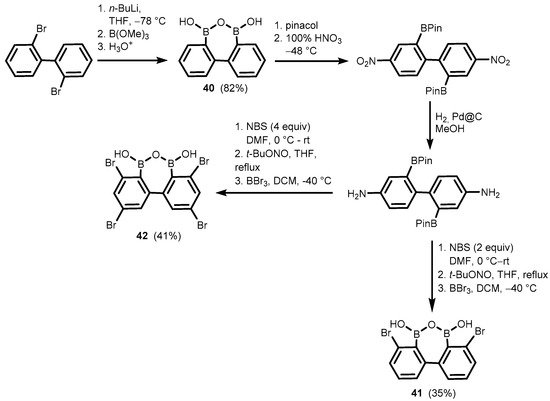
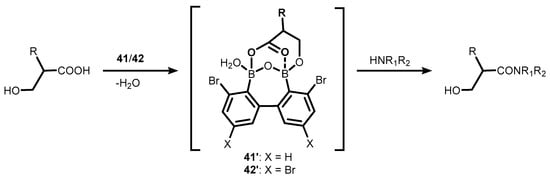
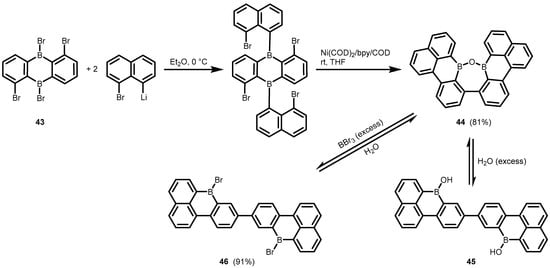
References
- Liu, C.T.; Tomsho, J.W.; Benkovic, S.J. The Unique Chemistry of Benzoxaboroles: Current and Emerging Applications in Biotechnology and Therapeutic Treatments. Bioorg. Med. Chem. 2014, 22, 4462–4473.
- Adamczyk-Woźniak, A.; Borys, K.M.; Sporzyński, A. Recent Developments in the Chemistry and Biological Applications of Benzoxaboroles. Chem. Rev. 2015, 115, 5224–5247.
- Yang, F.; Zhu, M.; Zhang, J.; Zhou, H. Synthesis of Biologically Active Boron-Containing Compounds. Med. Chem. Commun. 2018, 9, 201–211.
- Nocentini, A.; Supuran, C.T.; Winum, J.-Y. Benzoxaborole Compounds for Therapeutic Uses: A Patent Review (2010–2018). Expert Opin. Ther. Pat. 2018, 28, 493–504.
- Kaufmann, D.E.; Boese, R.; Scheer, A. 1,2-Bis(diisopropylamino)-1,2-dihydro-1,2-benzodiboret-ein erstes thermisch stabiles 1,2-Dihydro-1,2-diboret. Chem. Ber. 1994, 127, 2349–2351.
- Kaufmann, D. Borylierung von Arylsilanen, II Synthese und Reaktionen silylierter Dihalogenphenylborane. Chem. Ber. 1987, 120, 901–905.
- Durka, K.; Jarzembska, K.N.; Kamiński, R.; Luliński, S.; Serwatowski, J.; Woźniak, K. Nanotubular Hydrogen-Bonded Organic Framework Architecture of 1,2-Phenylenediboronic Acid Hosting Ice Clusters. Cryst. Growth Des. 2013, 13, 4181–4185.
- Durka, K.; Luliński, S.; Serwatowski, J.; Woźniak, K. Influence of Fluorination and Boronic Group Synergy on the Acidity and Structural Behavior of o-Phenylenediboronic Acids. Organometallics 2014, 33, 1608–1616.
- Adamczyk-Woźniak, A.; Cyrański, M.; Durka, K.; Gozdalik, J.; Klimentowska, P.; Rusiecki, R.; Sporzyński, A.; Zarzeczańska, D. Structure and Properties of 1,3-Phenylenediboronic Acid: Combined Experimental and Theoretical Investigations. Crystals 2019, 9, 109.
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Kubsik, M.; Nawara, K.; Witkowska, E.; Wiloch, M.; Luliński, S.; Waluk, J.; Głowacki, I.; et al. New Class of Easily-Synthesisable and Modifiable Organic Materials for Applications in Luminescent Devices. Dye. Pigm. 2017, 138, 267–277.
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Kubsik, M. Engineering of Solvatomorphs of the Luminescent Complex of ortho -Phenylenediboronic Acid and 8-Hydroxyquinoline. Cryst. Growth Des. 2017, 17, 6836–6851.
- Jarzembska, K.N.; Kamiński, R.; Durka, K.; Woźniak, K. Ground-State Charge-Density Distribution in a Crystal of the Luminescent Ortho-Phenylenediboronic Acid Complex with 8-Hydroxyquinoline. J. Phys. Chem. A 2018, 122, 4508–4520.
- Kutniewska, S.E.; Jarzembska, K.N.; Kamiński, R.; Stasyuk, A.J.; Gryko, D.T.; Cyrański, M.K. Structural, Energetic and Spectroscopic Studies of New Luminescent Complexes Based on 2-(2′-Hydroxyphenyl)Imidazo[1,2-a]Pyridines and 1,2-Phenylenediboronic Acid. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2018, 74, 725–737.
- Williams, V.C.; Piers, W.E.; Clegg, W.; Elsegood, M.R.J.; Collins, S.; Marder, T.B. New Bifunctional Perfluoroaryl Boranes. Synthesis and Reactivity of the ortho-Phenylene-Bridged Diboranes 1,2-[B(C6F5)2]2C6X4 (X = H, F). J. Am. Chem. Soc. 1999, 121, 3244–3245.
- Williams, V.C.; Irvine, G.J.; Piers, W.E.; Li, Z.; Collins, S.; Clegg, W.; Elsegood, M.R.J.; Marder, T.B. Novel Trityl Activators with New Weakly Coordinating Anions Derived from C6F4-1,2-[B(C6F5)2]2: Synthesis, Structures, and Olefin Polymerization Behavior. Organometallics 2000, 19, 1619–1621.
- Lewis, S.P.; Taylor, N.J.; Piers, W.E.; Collins, S. Isobutene Polymerization Using a Chelating Diborane Co-Initiator. J. Am. Chem. Soc. 2003, 125, 14686–14687.
- Chai, J.; Lewis, S.P.; Collins, S.; Sciarone, T.J.J.; Henderson, L.D.; Chase, P.A.; Irvine, G.J.; Piers, W.E.; Elsegood, M.R.J.; Clegg, W. Formation of Chelated Counteranions Using Lewis Acidic Diboranes: Relevance to Isobutene Polymerization. Organometallics 2007, 26, 5667–5679.
- Henderson, L.D.; Piers, W.E. Ion Pair Symmetrization in Metallocenium Cations Partnered with Diborane Derived Borate Counteranions. J. Organomet. Chem. 2007, 692, 4661–4668.
- Henderson, L.D.; Piers, W.E.; Irvine, G.J.; McDonald, R. Anion Stability in Stannylium, Oxonium, and Silylium Salts of the Weakly Coordinating Anion [C6F4-1,2-2(μ-OCH3)]−. Organometallics 2002, 21, 340–345.
- Lewis, S.P.; Chai, J.; Collins, S.; Sciarone, T.J.J.; Henderson, L.D.; Fan, C.; Parvez, M.; Piers, W.E. Isobutene Polymerization Using Chelating Diboranes: Polymerization in Aqueous Suspension and Hydrocarbon Solution. Organometallics 2009, 28, 249–263.
- Sgro, M.J.; Dömer, J.; Stephan, D.W. Stoichiometric CO2 Reductions Using a Bis-Borane-Based Frustrated Lewis Pair. Chem. Commun. 2012, 48, 7253.
- Letsinger, R.L.; Smith, J.M.; Gilpin, J.; MacLean, D.B. Organoboron Compounds. XX. Chemistry of Some 1-Naphthaleneboronic Acids with Substituents in the 8-Position. J. Org. Chem. 1965, 30, 807–812.
- Katz, H.E. Synthesis and Characterization of Novel 1H,3H-Naphth[1,8-cd][1,2,6]oxadiborins. J. Org. Chem. 1985, 50, 2575–2576.
- Scholz, A.S.; Massoth, J.G.; Bursch, M.; Mewes, J.-M.; Hetzke, T.; Wolf, B.; Bolte, M.; Lerner, H.-W.; Grimme, S.; Wagner, M. BNB-Doped Phenalenyls: Modular Synthesis, Optoelectronic Properties, and One-Electron Reduction. J. Am. Chem. Soc. 2020, 142, 11072–11083.
- Rzepa, H.S.; Arkhipenko, S.; Wan, E.; Sabatini, M.T.; Karaluka, V.; Whiting, A.; Sheppard, T.D. An Accessible Method for DFT Calculation of 11B NMR Shifts of Organoboron Compounds. J. Org. Chem. 2018, 83, 8020–8025.
- Watkinson, M.; Whiting, A.; McAuliffe, C.A. Synthesis of a Bis-Manganese Water Splitting Complex. J. Chem. Soc. Chem. Commun. 1994, 2141.
- Li, X.; Han, J.-W.; Zhang, Y.-X.; Wong, H.N.C. Palladium-Catalyzed Double Suzuki Reactions: Synthesis of Dibenzo[4,5:6,7]Cyclohepta[1,2,3-de]Naphthalenes. Asian J. Org. Chem. 2017, 6, 1876–1884.
- Kusukawa, T.; Mura, R.; Ooe, M.; Sumida, R.; Nakagawa, A. Recognition of Carboxylic Acids and Phosphonic Acids Using 1,8-Diphenylnaphthalene-Based Diguanidine. Tetrahedron 2021, 77, 131770.
- Yang, J.; Horst, M.; Werby, S.H.; Cegelski, L.; Burns, N.Z.; Xia, Y. Bicyclohexene-Peri-Naphthalenes: Scalable Synthesis, Diverse Functionalization, Efficient Polymerization, and Facile Mechanoactivation of Their Polymers. J. Am. Chem. Soc. 2020, 142, 14619–14626.
- IJpeij, E.G.; Beijer, F.H.; Arts, H.J.; Newton, C.; de Vries, J.G.; Gruter, G.-J.M. A Suzuki Coupling Based Route to 2,2′-Bis(2-indenyl)biphenyl Derivatives. J. Org. Chem. 2002, 67, 169–176.
- Das, A.; Hübner, A.; Weber, M.; Bolte, M.; Lerner, H.-W.; Wagner, M. 9-H-9-Borafluorene Dimethyl Sulfide Adduct: A Product of a Unique Ring-Contraction Reaction and a Useful Hydroboration Reagent. Chem. Commun. 2011, 47, 11339–11341.
- Shimada, N.; Hirata, M.; Koshizuka, M.; Ohse, N.; Kaito, R.; Makino, K. Diboronic Acid Anhydrides as Effective Catalysts for the Hydroxy-Directed Dehydrative Amidation of Carboxylic Acids. Org. Lett. 2019, 21, 4303–4308.
- Shimada, N.; Takahashi, N.; Ohse, N.; Koshizuka, M.; Makino, K. Synthesis of Weinreb Amides Using Diboronic Acid Anhydride-Catalyzed Dehydrative Amidation of Carboxylic Acids. Chem. Commun. 2020, 56, 13145–13148.
- Shimada, N.; Ohse, N.; Takahashi, N.; Urata, S.; Koshizuka, M.; Makino, K. Direct Synthesis of N-Protected Serine- and Threonine-Derived Weinreb Amides via Diboronic Acid Anhydride-Catalyzed Dehydrative Amidation: Application to the Concise Synthesis of Garner’s Aldehyde. Synlett 2021, 32, 1024–1028.
- Koshizuka, M.; Makino, K.; Shimada, N. Diboronic Acid Anhydride-Catalyzed Direct Peptide Bond Formation Enabled by Hydroxy-Directed Dehydrative Condensation. Org. Lett. 2020, 22, 8658–8664.
- Radtke, J.; Schickedanz, K.; Bamberg, M.; Menduti, L.; Schollmeyer, D.; Bolte, M.; Lerner, H.-W.; Wagner, M. Selective Access to Either a Doubly Boron-Doped Tetrabenzopentacene or an Oxadiborepin from the Same Precursor. Chem. Sci. 2019, 10, 9017–9027.

