 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Hsu-Shan Huang | + 2298 word(s) | 2298 | 2021-09-16 08:09:04 | | | |
| 2 | Jason Zhu | Meta information modification | 2298 | 2021-09-17 04:54:19 | | | | |
| 3 | Jason Zhu | Meta information modification | 2298 | 2021-09-18 12:14:42 | | | | |
| 4 | Jason Zhu | -23 word(s) | 2275 | 2021-10-26 09:44:36 | | |
Video Upload Options
Glioblastoma (GBM) is one of the most aggressive brain malignancies with high incidences of developing treatment resistance, resulting in poor prognoses. Glioma stem cell (GSC)-derived exosomes are important players that contribute to GBM tumorigenesis and aggressive properties.
1. Introduction
Despite advancements in the knowledge and understanding of the mechanisms involved in the tumor biology of glioblastoma (GBM) and various treatment modalities over the last few decades, GBM remains one of the deadliest and the most common primary brain tumors, with tremendously poor prognoses[1]. The success of clinical trials of new chemotherapies and standard therapies has been disappointing[2] due to several factors including the drug delivery limiting features of blood–brain barrier (BBB), GBM immune-suppressive microenvironments, and structural fragility of the brain[3]. Furthermore, the existence of glioma-initiating cells (GICs) a type of glioma stem cell (GSC), mediates treatment failure, tumor recurrence, and invasive phenotypes of GBM [4]. As a carrier of oncogenes/proteins and other genetic information, exosomes are involved in the conversion of non-GSCs to GSCs and participate in stabilizing the GSC phenotypic integrity[5]. GBM is also characterized by complex and heterogeneous genotypes which limit the efficacy of drugs that target specific oncogenic signaling axes[6]. Thus, developing a multi-oncotarget treatment modality that targets GSCs and exosomes may improve the devastating prognoses of GBM.
The phosphatidylinositol-4,5-biphosphate 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway has emerged as one of the most deregulated oncogenic pathways that contribute to aggressive phenotypes, drug resistance, and poor prognoses in GBM patients[7]. However, due to several limiting factors [8][9][10][11], efforts to target this signaling axis failed to improve the prognoses of GBM patients [8]. Importantly, the inability of mTOR inhibitors to target mTORC2 epitomizes another major clinical limitation of targeted therapy [12] Therefore, targeting mTORC2 would overcome the limitations of mTORC1 inhibitors and provide a sound therapeutic strategy for GBM. This is supported by preclinical evidence of the critical roles of mTORC2 in GBM biology and the extenuating effect of targeting mTORC2 on GBM growth, invasive phenotypes, and drug resistance[13][14][15][16], thus paving the way for personalized and targeted therapy.
Cyclin-dependent kinases (CDKs) are members of the serine/threonine protein kinase family that regulates cell division and transcription . Unrestricted cell cycle progression and high cellular growth due to aberrant CDK4/6 signaling have been identified as hallmarks of astrocytic tumorigenesis and glioma progression in most GBM cases [17][18][19]. Hyper-expression of CDK4/6 was also documented in several other cancer types [20][21][22][23]. Although mono-therapeutic inhibitors that target CDK4/6 signaling pathways have been developed[24][25][26], their efficacy in GBM remains disappointing [18], thus accentuating the need for synergistic contributions from other agents.
Summing up the above literature with the clinical data from The Cancer Genome Atlas (TCGA) database strongly suggests that CDK6/mTOR/STAT3 overexpression is correlated with a high glioma grade, lower survival, and poor prognosis in glioblastoma patients [18][23]. The presence of GSCs and oncogene delivery features of exosomes [5], together with aberrations of CDK6/mTOR/STAT3 oncogenic pathways, concomitantly contribute to aggressive phenotypes and the failure of therapeutic strategies against GBM[25]. New therapeutic strategies are urgently needed to address all the challenges mentioned above to improve patients’ survival.
Palbociclib is an oral selective inhibitor of CDK4/6, which leads to phosphorylation of RB1 and cell-cycle arrest [26]. RB1 status, therefore, becomes a determinant of tumor sensitivity to palbociclib therapy. Disappointedly, about 11% of GBM show complete loss of RB1 transcript expression [27], rendering them resistant to palbociclib. Clinical studies have also demonstrated that CDK4/6 inhibitor alone showed sub-optimal efficacy for recurrent glioblastoma [29][30][31]. Thus, in combination with other therapies, palbociclib has been vigorously tested and proven effective in some patients [28][30][31][32][33][34][35][36]. Specifically, mTOR inhibitor with palbociclib showed increased efficacy against GBM [37][38]. In addition, there are several ongoing trials testing combinations of palbociclib with immunotherapy, including avelumab and pembrolizumab (NCT02778685; NCT02779751; and NCT03147287) [39]. Collectively, these findings strongly suggested that targeting mTOR/CDK6 associated signaling is a potential new target for developing GBM therapeutics.
Anthraquinone-derived heterocyclic scaffolds have been explored for drug design, discovery, and development[40], and drug candidates from this class have demonstrated antitumor activities in various studies [41]. GBM-N019 is a novel member of a series of anthraquinone-derived, tetraheterocylic azathioxanthone derivatives. Herein, we demonstrated for the first time through a series of in vitro and in vivo studies that GBM-N019 significantly compromised the viability and tumorigenic features of GBM cells via downregulation of STAT3, Akt, mTOR, nuclear factor (NF)-κB, and CDK6 signaling networks. We also found that GBM-N019 halted the exosomal cargo delivery of Akt, mTOR, p-mTOR, and RAB27A, and attenuated the tumorsphere-derived exosomes (exosphere; Exosp) mediated drug resistance and aggressive phenotypes of GBM.
2. mTOR, STAT3, and CDK6 Are Key Oncogenic Signatures of Disease Progression, Therapy Failure, and Poor Prognosis in GBM Patients
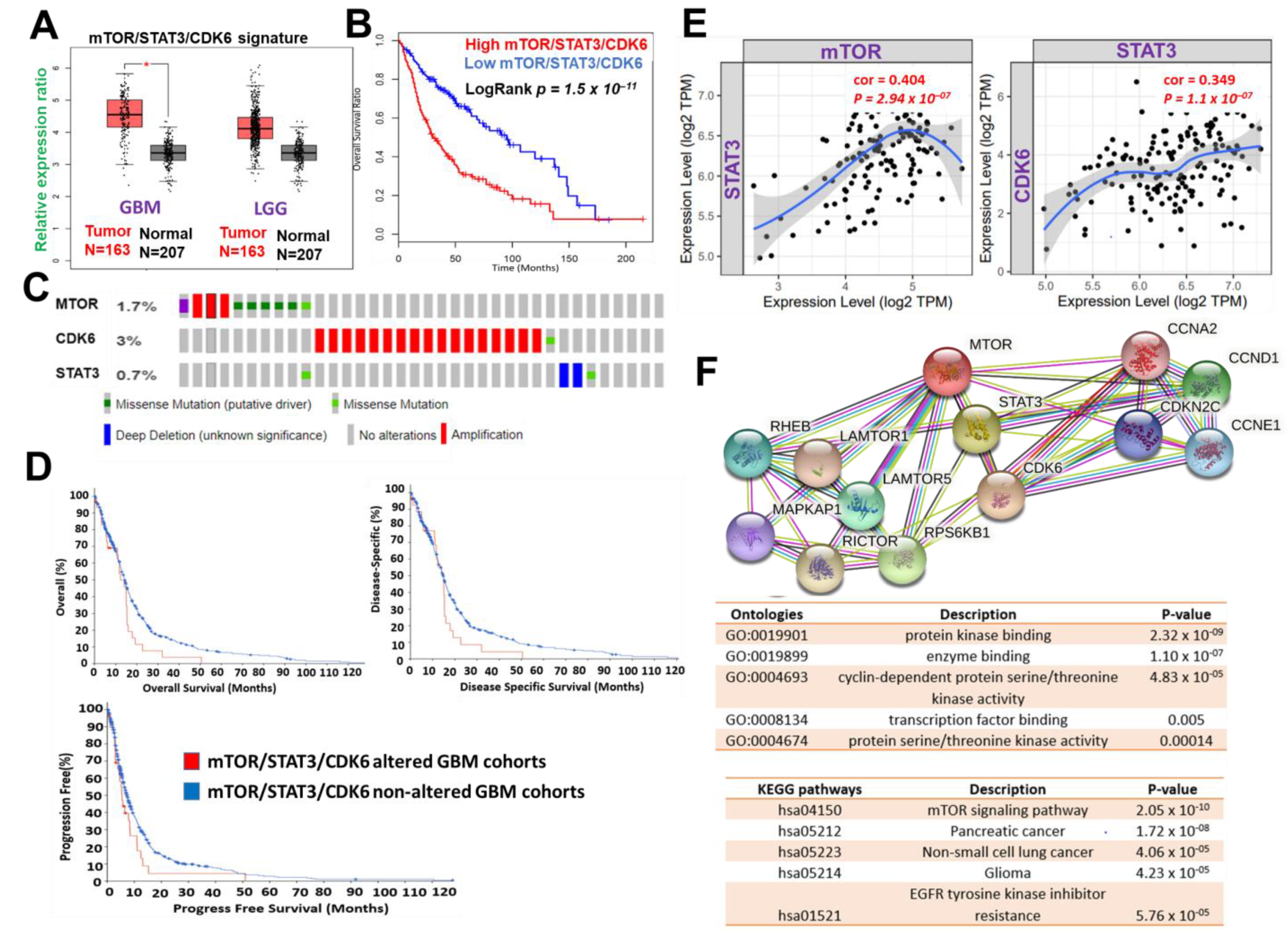 Figure 1. mTOR, STAT3 and CDK6 is an important oncogenic signature of disease progression, therapy failure, and poor prognosis in GBM patients. (A) Cumulative mTOR, STAT3, and CDK6 expression levels in the patients with GBM and low-grade glioma (LGG) from TCGA and GTEx datasets. (B) Kaplan–Meier plots of the cumulative survival of GBM patients. (C) Frequency of genetic alterations of mTOR, STAT3 and CDK6 signature in glioblastoma (TCGA, PanCancer Atlas) dataset. (D) Kaplan–Meier plots of the overall survival, disease-specific, and disease progressive free survival of GBM cohorts with genetically altered mTOR, STAT3 and CDK6 signature. (E) Correlation analysis of the expression between STAT3 and mTOR (left) and between CDK6 and STAT3 (right) from GBM TCGA databases. Both p-values and Spearman’s rank correlation coefficient (cor) are indicated. (F) Protein–protein interaction networks of mTOR, STAT3 and CDK6 signature (upper panel) and the enriched KEGG pathways and gene ontologies in the mTOR, STAT3 and CDK6 clustering network.
Figure 1. mTOR, STAT3 and CDK6 is an important oncogenic signature of disease progression, therapy failure, and poor prognosis in GBM patients. (A) Cumulative mTOR, STAT3, and CDK6 expression levels in the patients with GBM and low-grade glioma (LGG) from TCGA and GTEx datasets. (B) Kaplan–Meier plots of the cumulative survival of GBM patients. (C) Frequency of genetic alterations of mTOR, STAT3 and CDK6 signature in glioblastoma (TCGA, PanCancer Atlas) dataset. (D) Kaplan–Meier plots of the overall survival, disease-specific, and disease progressive free survival of GBM cohorts with genetically altered mTOR, STAT3 and CDK6 signature. (E) Correlation analysis of the expression between STAT3 and mTOR (left) and between CDK6 and STAT3 (right) from GBM TCGA databases. Both p-values and Spearman’s rank correlation coefficient (cor) are indicated. (F) Protein–protein interaction networks of mTOR, STAT3 and CDK6 signature (upper panel) and the enriched KEGG pathways and gene ontologies in the mTOR, STAT3 and CDK6 clustering network.3. mTOR, STAT3, and CDK6 Are Druggable Targets for a Novel Drug-like Multitarget Small Molecule (GBM-N019)
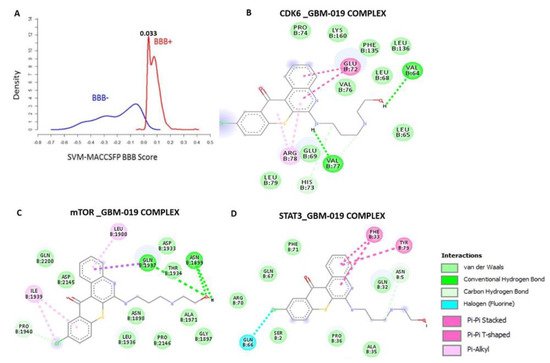
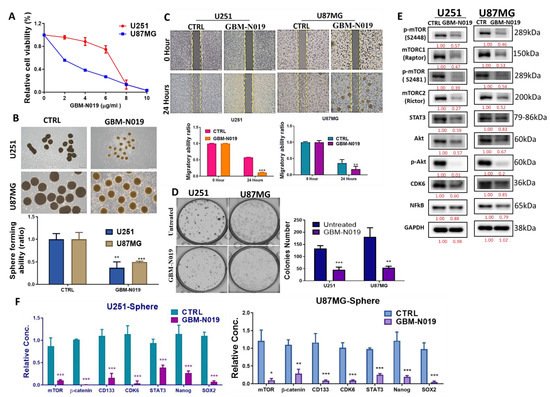
4. GBM-N019 Curbed the Viability and Tumorigenic Features of GBM Cells via Downregulation of NF-κB/Akt/mTOR, STAT3, and CDK6 Signaling In Vitro
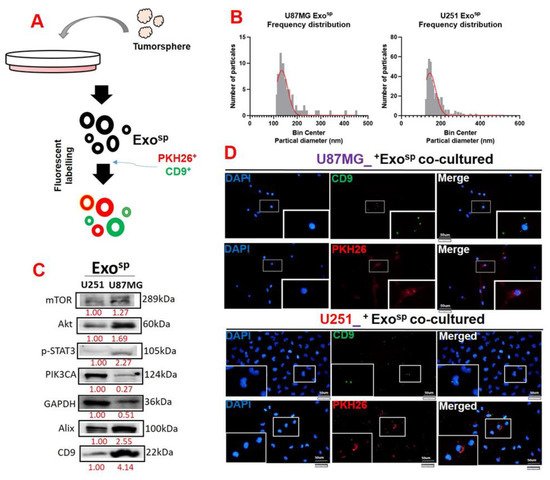
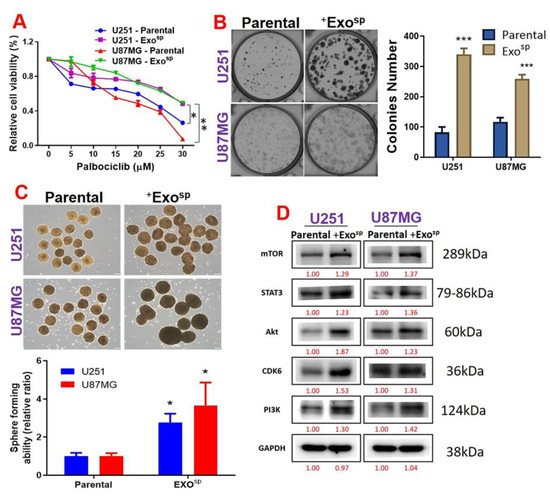
5. Tumorsphere-Derived Exosomal Cargo of Oncogenes Mediated Treatment Resistance and Aggressive Phenotypes of GBM
6. Conclusions
References
- Hess, K.R.; Broglio, K.R.; Bondy, M.L. Adult glioma incidence trends in the United States, 1977–2000. Cancer 2004, 101, 2293–2299. [Google Scholar] [CrossRef]
- Sauter, E.R. Cancer prevention and treatment using combination therapy with natural compounds. Expert Rev. Clin. Pharm. 2020, 13, 265–285. [Google Scholar] [CrossRef]
- Moradimotlagh, A.; Arefian, E.; Rezazadeh Valojerdi, R.; Ghaemi, S.; Jamshidi Adegani, F.; Soleimani, M. MicroRNA-129 Inhibits Glioma Cell Growth by Targeting CDK4, CDK6, and MDM2. Mol. Ther Nucleic Acids 2020, 19, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Linhares, P.; Carvalho, B.; Vaz, R.; Costa, B.M. Glioblastoma: Is There Any Blood Biomarker with True Clinical Relevance? Int. J. Mol. Sci. 2020, 21, 5809. [Google Scholar] [CrossRef] [PubMed]
- Stella, M.; Falzone, L.; Caponnetto, A.; Gattuso, G.; Barbagallo, C.; Battaglia, R.; Mirabella, F.; Broggi, G.; Altieri, R.; Certo, F.; et al. Serum Extracellular Vesicle-Derived circHIPK3 and circSMARCA5 Are Two Novel Diagnostic Biomarkers for Glioblastoma Multiforme. Pharmaceuticals 2021, 14, 618. [Google Scholar] [CrossRef] [PubMed]
- Silantyev, A.S.; Falzone, L.; Libra, M.; Gurina, O.I.; Kardashova, K.S.; Nikolouzakis, T.K.; Nosyrev, A.E.; Sutton, C.W.; Mitsias, P.D.; Tsatsakis, A. Current and Future Trends on Diagnosis and Prognosis of Glioblastoma: From Molecular Biology to Proteomics. Cells 2019, 8, 863. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wu, C.; Chen, N.; Gu, H.; Yen, A.; Cao, L.; Wang, E.; Wang, L. PI3K/Akt/mTOR signaling pathway and targeted therapy for glioblastoma. Oncotarget 2016, 7, 33440–33450. [Google Scholar] [CrossRef] [PubMed]
- Galanis, E.; Buckner, J.C.; Maurer, M.J.; Kreisberg, J.I.; Ballman, K.; Boni, J.; Peralba, J.M.; Jenkins, R.B.; Dakhil, S.R.; Morton, R.F.; et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: A North Central Cancer Treatment Group Study. J. Clin. Oncol. 2005, 23, 5294–5304. [Google Scholar] [CrossRef] [PubMed]
- Akhavan, D.; Cloughesy, T.F.; Mischel, P.S. mTOR signaling in glioblastoma: Lessons learned from bench to bedside. Neuro-Oncology 2010, 12, 882–889. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.-f.; Wang, J.; Shao, W.; Wu, C.-p.; Chen, Z.-p.; To, S.-s.T.; Li, W.-p. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 100. [Google Scholar] [CrossRef]
- Reilly, K.E.; Rojo, F.; She, Q.-B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR Inhibition Induces Upstream Receptor Tyrosine Kinase Signaling and Activates Akt. Cancer Res. 2006, 66, 1500. [Google Scholar] [CrossRef]
- Choo, A.Y.; Yoon, S.-O.; Kim, S.G.; Roux, P.P.; Blenis, J. Rapamycin differentially inhibits S6Ks and 4E-BP1 to mediate cell-type-specific repression of mRNA translation. Proc. Natl. Acad. Sci. USA 2008, 105, 17414–17419. [Google Scholar] [CrossRef] [PubMed]
- Zaytseva, Y.Y.; Valentino, J.D.; Gulhati, P.; Mark Evers, B. mTOR inhibitors in cancer therapy. Cancer Lett. 2012, 319, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gini, B.; Zanca, C.; Guo, D.; Matsutani, T.; Masui, K.; Ikegami, S.; Yang, H.; Nathanson, D.; Villa, G.R.; Shackelford, D.; et al. The mTOR Kinase Inhibitors, CC214-1 and CC214-2, Preferentially Block the Growth of EGFRvIII-Activated Glioblastomas. Clin. Cancer Res. 2013, 19, 5722. [Google Scholar] [CrossRef]
- Mortensen, D.S.; Sapienza, J.; Lee, B.G.S.; Perrin-Ninkovic, S.M.; Harris, R.; Shevlin, G.; Parnes, J.S.; Whitefield, B.; Hickman, M.; Khambatta, G.; et al. Use of core modification in the discovery of CC214-2, an orally available, selective inhibitor of mTOR kinase. Bioorg. Med. Chem. Lett. 2013, 23, 1588–1591. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef]
- Tadesse, S.; Yu, M.; Kumarasiri, M.; Le, B.T.; Wang, S. Targeting CDK6 in cancer: State of the art and new insights. Cell Cycle 2015, 14, 3220–3230. [Google Scholar] [CrossRef]
- Schröder, L.B.; McDonald, K.L. CDK4/6 inhibitor PD0332991 in glioblastoma treatment: Does it have a future? Front. Oncol. 2015, 5, 259. [Google Scholar] [CrossRef]
- Timmermann, S.; Hinds, P.W.; Münger, K. Elevated activity of cyclin-dependent kinase 6 in human squamous cell carcinoma lines. Cell Growth Differ. 1997, 8, 361–370. [Google Scholar]
- Lee, K.-H.; Lotterman, C.; Karikari, C.; Omura, N.; Feldmann, G.; Habbe, N.; Goggins, M.G.; Mendell, J.T.; Maitra, A. Epigenetic Silencing of MicroRNA miR-107 Regulates Cyclin-Dependent Kinase 6 Expression in Pancreatic Cancer. Pancreatology 2009, 9, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zheng, L.; Yu, Z.; Liao, G.; Lu, L.; Xu, R.; Zhao, Z.; Chen, G. Increased cyclin-dependent kinase 6 expression in bladder cancer Corrigendum in/ol/5/6/1979. Oncol. Lett. 2012, 4, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, A.; Sicinski, P.; Hinds, P.W. Cyclins and cdks in development and cancer: A perspective. Oncogene 2005, 24, 2909–2915. [Google Scholar] [CrossRef] [PubMed]
- Lawal, B.; Lin, L.-C.; Lee, J.-C.; Chen, J.-H.; Bekaii-Saab, T.S.; Wu, A.T.H.; Ho, C.-L. Multi-Omics Data Analysis of Gene Expressions and Alterations, Cancer-Associated Fibroblast and Immune Infiltrations, Reveals the Onco-Immune Prognostic Relevance of STAT3/CDK2/4/6 in Human Malignancies. Cancers 2021, 13, 954. [Google Scholar] [CrossRef]
- Sarcar, B.; Kahali, S.; Prabhu, A.H.; Shumway, S.D.; Xu, Y.; Demuth, T.; Chinnaiyan, P. Targeting Radiation-Induced G2 Checkpoint Activation with the Wee-1 Inhibitor MK-1775 in Glioblastoma Cell Lines. Mol. Cancer Ther. 2011, 10, 2405. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, F.A.; Hossain, M.K.; Mostofa, A.G.M.; Akbor, M.M.; Bin Sayeed, M.S. Therapeutic Potential of Thymoquinone in Glioblastoma Treatment: Targeting Major Gliomagenesis Signaling Pathways. BioMed Res. Int. 2018, 2018, 4010629. [Google Scholar] [CrossRef]
- Clark, A.S.; Karasic, T.B.; DeMichele, A.; Vaughn, D.J.; O’Hara, M.; Perini, R.; Zhang, P.; Lal, P.; Feldman, M.; Gallagher, M.; et al. Palbociclib (PD0332991)—a Selective and Potent Cyclin-Dependent Kinase Inhibitor: A Review of Pharmacodynamics and Clinical Development. JAMA Oncol. 2016, 2, 253–260. [Google Scholar] [CrossRef]
- Goldhoff, P.; Clarke, J.; Smirnov, I.; Berger, M.S.; Prados, M.D.; James, C.D.; Perry, A.; Phillips, J.J. Clinical stratification of glioblastoma based on alterations in retinoblastoma tumor suppressor protein (RB1) and association with the proneural subtype. J. Neuropathol. Exp. Neurol. 2012, 71, 83–89. [Google Scholar] [CrossRef]
- Whittaker, S.; Madani, D.; Joshi, S.; Chung, S.A.; Johns, T.; Day, B.; Khasraw, M.; McDonald, K.L. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017, 3, 17033. [Google Scholar] [CrossRef]
- Tien, A.-C.; Li, J.; Bao, X.; Derogatis, A.; Kim, S.; Mehta, S.; Sanai, N. A Phase 0 Trial of Ribociclib in Recurrent Glioblastoma Patients Incorporating a Tumor Pharmacodynamic- and Pharmacokinetic-Guided Expansion Cohort. Clin. Cancer Res. 2019, 25, 5777–5786. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Parikh, M.; Phillips, J.J.; James, C.D.; Molinaro, A.M.; Butowski, N.A.; Clarke, J.L.; Oberheim-Bush, N.A.; Chang, S.M.; Berger, M.S.; et al. Phase-2 trial of palbociclib in adult patients with recurrent RB1-positive glioblastoma. J. Neuro-oncol. 2018, 140, 477–483. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda-Sánchez, J.M.; Gil-Gil, M.; Alonso-García, M.; Vaz Salgado, M.Á.; Vicente, E.; Mesía Barroso, C.; Rodríguez Sánchez, Á.; Durán, G.; De Las Peñas, R.; Muñoz-Langa, J.; et al. Phase II Trial of Palbociclib in Recurrent Retinoblastoma-Positive Anaplastic Oligodendroglioma: A Study from the Spanish Group for Research in Neuro-Oncology (GEINO). Target. Oncol. 2020, 15, 613–622. [Google Scholar] [CrossRef]
- McShane, T.M.; Wolfe, T.A.; Ryan, J.C. Updates on managing advanced breast cancer with palbociclib combination therapy. Ther. Adv. Med. Oncol. 2018, 10, 1758835918793849. [Google Scholar] [CrossRef]
- Li, M.; Xiao, A.; Floyd, D.; Olmez, I.; Lee, J.; Godlewski, J.; Bronisz, A.; Bhat, K.P.L.; Sulman, E.P.; Nakano, I.; et al. CDK4/6 inhibition is more active against the glioblastoma proneural subtype. Oncotarget 2017, 8, 55319–55331. [Google Scholar] [CrossRef]
- Zhang, J.; Bu, X.; Wang, H.; Zhu, Y.; Geng, Y.; Nihira, N.T.; Tan, Y.; Ci, Y.; Wu, F.; Dai, X.; et al. Cyclin D–CDK4 kinase destabilizes PD-L1 via cullin 3–SPOP to control cancer immune surveillance. Nature 2018, 553, 91–95. [Google Scholar] [CrossRef]
- Hashizume, R.; Zhang, A.; Mueller, S.; Prados, M.D.; Lulla, R.R.; Goldman, S.; Saratsis, A.M.; Mazar, A.P.; Stegh, A.H.; Cheng, S.Y.; et al. Inhibition of DNA damage repair by the CDK4/6 inhibitor palbociclib delays irradiated intracranial atypical teratoid rhabdoid tumor and glioblastoma xenograft regrowth. Neuro. Oncol. 2016, 18, 1519–1528. [Google Scholar] [CrossRef]
- Michaud, K.; Solomon, D.A.; Oermann, E.; Kim, J.S.; Zhong, W.Z.; Prados, M.D.; Ozawa, T.; James, C.D.; Waldman, T. Pharmacologic inhibition of cyclin-dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res. 2010, 70, 3228–3238. [Google Scholar] [CrossRef]
- Liu, S.; Tang, Y.; Yuan, X.; Yuan, D.; Liu, J.; Li, B.; Li, Y. Inhibition of Rb and mTOR signaling associates with synergistic anticancer effect of palbociclib and erlotinib in glioblastoma cells. Invest. New Drugs 2018, 36, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Olmez, I.; Brenneman, B.; Xiao, A.; Serbulea, V.; Benamar, M.; Zhang, Y.; Manigat, L.; Abbas, T.; Lee, J.; Nakano, I.; et al. Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin. Cancer Res. 2017, 23, 6958–6968. [Google Scholar] [CrossRef] [PubMed]
- Anurag, M.; Haricharan, S.; Ellis, M.J. CDK4/6 Inhibitor Biomarker Research: Are We Barking Up the Wrong Tree? Clin. Cancer Res. 2020, 26, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.-C.; Wu, C.-L.; Lee, C.-C.; Chen, C.-L.; Yu, D.-S.; Huang, H.-S. Structure-based hybridization, synthesis and biological evaluation of novel tetracyclic heterocyclic azathioxanthone analogues as potential antitumor agents. Eur. J. Med. Chem. 2015, 103, 615–627.
- Lawal, B.; Liu, Y.-L.; Mokgautsi, N.; Khedkar, H.; Sumitra, M.R.; Wu, A.T.H.; Huang, H.-S. Pharmacoinformatics and Preclinical Studies of NSC765690 and NSC765599, Potential STAT3/CDK2/4/6 Inhibitors with Antitumor Activities against NCI60 Human Tumor Cell Lines. Biomedicines 2021, 9, 92.

