Glioblastoma (GBM) is one of the most aggressive brain malignancies with high incidences of developing treatment resistance, resulting in poor prognoses. Glioma stem cell (GSC)-derived exosomes are important players that contribute to GBM tumorigenesis and aggressive properties.
1. Introduction
Despite advancements in the knowledge and understanding of the mechanisms involved in the tumor biology of glioblastoma (GBM) and various treatment modalities over the last few decades, GBM remains one of the deadliest and the most common primary brain tumors, with tremendously poor prognoses [1]. The success of clinical trials of new chemotherapies and standard therapies has been disappointing [2] due to several factors including the drug delivery limiting features of blood–brain barrier (BBB), GBM immune-suppressive microenvironments, and structural fragility of the brain [3]. Furthermore, the existence of glioma-initiating cells (GICs) a type of glioma stem cell (GSC), mediates treatment failure, tumor recurrence, and invasive phenotypes of GBM [4]. As a carrier of oncogenes/proteins and other genetic information, exosomes are involved in the conversion of non-GSCs to GSCs and participate in stabilizing the GSC phenotypic integrity [5]. GBM is also characterized by complex and heterogeneous genotypes which limit the efficacy of drugs that target specific oncogenic signaling axes [6]. Thus, developing a multi-oncotarget treatment modality that targets GSCs and exosomes may improve the devastating prognoses of GBM.
The phosphatidylinositol-4,5-biphosphate 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway has emerged as one of the most deregulated oncogenic pathways that contribute to aggressive phenotypes, drug resistance, and poor prognoses in GBM patients [7]. However, due to several limiting factors [8,9,10,11], efforts to target this signaling axis failed to improve the prognoses of GBM patients [8]. Importantly, the inability of mTOR inhibitors to target mTORC2 epitomizes another major clinical limitation of targeted therapy [12]. Therefore, targeting mTORC2 would overcome the limitations of mTORC1 inhibitors and provide a sound therapeutic strategy for GBM. This is supported by preclinical evidence of the critical roles of mTORC2 in GBM biology and the extenuating effect of targeting mTORC2 on GBM growth, invasive phenotypes, and drug resistance [13,14,15], thus paving the way for personalized and targeted therapy.
Cyclin-dependent kinases (CDKs) are members of the serine/threonine protein kinase family that regulates cell division and transcription [16,17]. Unrestricted cell cycle progression and high cellular growth due to aberrant CDK4/6 signaling have been identified as hallmarks of astrocytic tumorigenesis and glioma progression in most GBM cases [18]. Hyper-expression of CDK4/6 was also documented in several other cancer types [19,20,21,22,23]. Although mono-therapeutic inhibitors that target CDK4/6 signaling pathways have been developed [24,25], their efficacy in GBM remains disappointing [18], thus accentuating the need for synergistic contributions from other agents.
Summing up the above literature with the clinical data from The Cancer Genome Atlas (TCGA) database strongly suggests that CDK6/mTOR/STAT3 overexpression is correlated with a high glioma grade, lower survival, and poor prognosis in glioblastoma patients [18,23]. The presence of GSCs and oncogene delivery features of exosomes [5], together with aberrations of CDK6/mTOR/STAT3 oncogenic pathways, concomitantly contribute to aggressive phenotypes and the failure of therapeutic strategies against GBM [25]. New therapeutic strategies are urgently needed to address all the challenges mentioned above to improve patients’ survival.
Palbociclib is an oral selective inhibitor of CDK4/6, which leads to phosphorylation of RB1 and cell-cycle arrest [26]. RB1 status, therefore, becomes a determinant of tumor sensitivity to palbociclib therapy. Disappointedly, about 11% of GBM show complete loss of RB1 transcript expression [27], rendering them resistant to palbociclib [28]. Clinical studies have also demonstrated that CDK4/6 inhibitor alone showed sub-optimal efficacy for recurrent glioblastoma [29,30,31]. Thus, in combination with other therapies, palbociclib has been vigorously tested and proven effective in some patients [28,31,32,33,34,35,36]. Specifically, mTOR inhibitor with palbociclib showed increased efficacy against GBM [37,38]. In addition, there are several ongoing trials testing combinations of palbociclib with immunotherapy, including avelumab and pembrolizumab (NCT02778685; NCT02779751; and NCT03147287) [39]. Collectively, these findings strongly suggested that targeting mTOR/CDK6 associated signaling is a potential new target for developing GBM therapeutics.
Anthraquinone-derived heterocyclic scaffolds have been explored for drug design, discovery, and development [40], and drug candidates from this class have demonstrated antitumor activities in various studies [41,42,43,44,45]. GBM-N019 is a novel member of a series of anthraquinone-derived, tetraheterocylic azathioxanthone derivatives. Herein, we demonstrated for the first time through a series of in vitro and in vivo studies that GBM-N019 significantly compromised the viability and tumorigenic features of GBM cells via downregulation of STAT3, Akt, mTOR, nuclear factor (NF)-κB, and CDK6 signaling networks. We also found that GBM-N019 halted the exosomal cargo delivery of Akt, mTOR, p-mTOR, and RAB27A, and attenuated the tumorsphere-derived exosomes (exosphere; Exosp) mediated drug resistance and aggressive phenotypes of GBM.
2. Results
2.1. mTOR, STAT3, and CDK6 Are Key Oncogenic Signatures of Disease Progression, Therapy Failure, and Poor Prognosis in GBM Patients
The use of bioinformatics databases and software for biomarkers and drug target predictions have greatly enhanced the process of drug design and development [
67]. Our analysis of clinical data from the TCGA database indicated that mTOR, STAT3, and CDK6 are collectively hyper-expressed in both low-grade glioma and GBM cohorts (
Figure 1A). These hyper-expressions were also found to correspond with the poor overall survival of cancer patients (
Figure 1B). The clinical data of glioblastoma cohorts in TCGA, PanCancer Atlas and found that alterations in mTOR, STAT3, and CDK6 constituted 5% of the genetic alteration occurring in GBM patients (
Figure 1C). These genetic alterations were found to be associated with shorter overall survival, disease-specific survival, and progression-free survival of the cohorts (
Figure 1D). Additionally, we identified a strong correlation between expressions of mTOR and STAT3 (
P = 2.95 × 10
−7, r = 0.405) and STAT3, as well as between STAT3 and CDK6 (
P = 1.1 × 10
−7, r = 0.349) (
Figure 1E). Analysis of functional protein association networks of mTOR, STAT3, and CDK6 identified (PPI enrichment p-value: 4.17 × 10
−8) CCNA2, CCNE1, CCND3, CCND1, Rictor, Raptor, MLST8, RHEB, and FKBP1A to be functional partners associated with their expressions and oncogenic properties in GBM (
Figure 1F). The PPI networks of mTOR, STAT3, and CDK6 were enriched in cancer-associated KEGG pathways and ontologies, among which cyclin-dependent protein serine/threonine kinase activity, transcription factor binding, mTOR signaling pathway, pancreatic cancer, non-small cell lung cancer, glioma, EGFR tyrosine kinase inhibitor resistance were those enriched (
Figure 2F lower panel). Collectively, this preliminary computational study revealed that mTOR, STAT3, and CDK6 is an important oncogenic signature that is associated with disease progression, therapy failure, and poor prognosis of GBM cohorts.
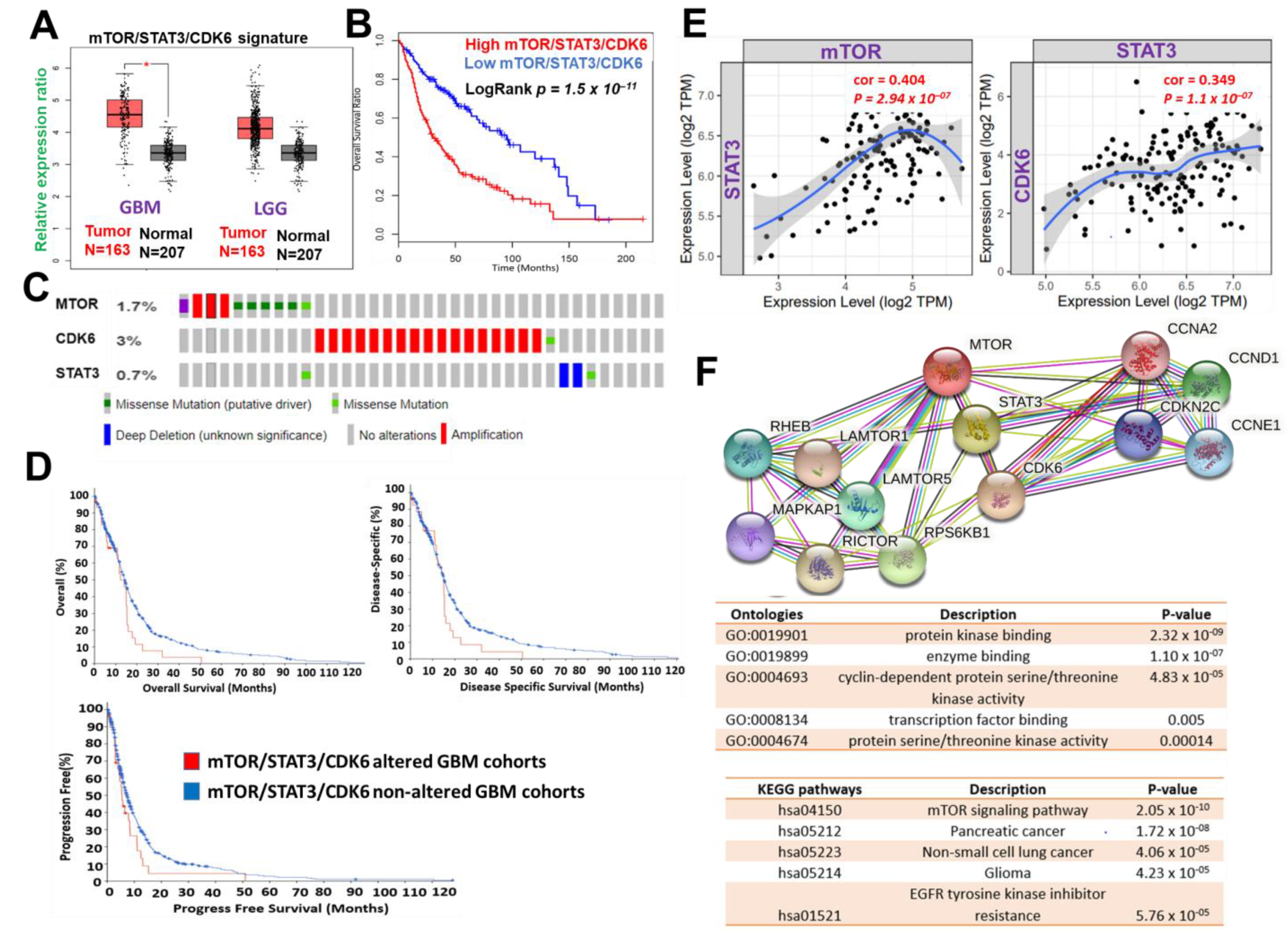 Figure 1.
Figure 1. mTOR, STAT3 and CDK6 is an important oncogenic signature of disease progression, therapy failure, and poor prognosis in GBM patients. (
A) Cumulative mTOR, STAT3, and CDK6 expression levels in the patients with GBM and low-grade glioma (LGG) from TCGA and GTEx datasets. (
B) Kaplan–Meier plots of the cumulative survival of GBM patients. (
C) Frequency of genetic alterations of mTOR, STAT3 and CDK6 signature in glioblastoma (TCGA, PanCancer Atlas) dataset. (
D) Kaplan–Meier plots of the overall survival, disease-specific, and disease progressive free survival of GBM cohorts with genetically altered mTOR, STAT3 and CDK6 signature. (
E) Correlation analysis of the expression between STAT3 and mTOR (
left) and between CDK6 and STAT3 (
right) from GBM TCGA databases. Both
p-values and Spearman’s rank correlation coefficient (cor) are indicated. (
F) Protein–protein interaction networks of mTOR, STAT3 and CDK6 signature (upper panel) and the enriched KEGG pathways and gene ontologies in the mTOR, STAT3 and CDK6 clustering network.
.2. mTOR, STAT3, and CDK6 Are Druggable Targets for a Novel Drug-like Multitarget Small Molecule (GBM-N019)
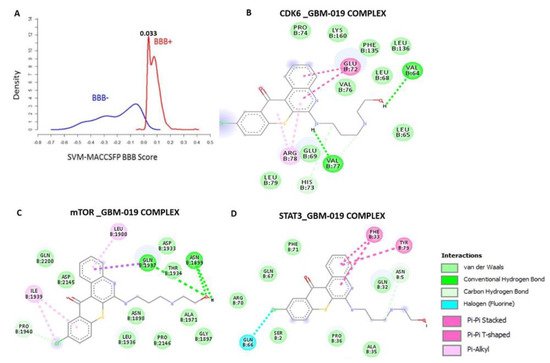
Computational study revealed that the GBM-N019 is BBB permeant (Figure 2A), it has acceptable absorption, distribution, metabolism, and excretion (ADME) characteristics and met all criteria of Lipinski’s rule of five. Furthermore, with computer-based drug target predictions and using GBM-N019 as a query molecule, we identified a number of targetable proteins, most of which were previously implicated in the poor prognosis of GBM. Among these predicted target proteins, enzymes (33.3%), kinases (26.7%), proteases (13.3%), electrochemical transporters (13.3%), and family A G protein-coupled receptors (13.3%) were the most repartitioned targeted classes . Specifically, three members of cyclin-dependent kinases (CDK4, -6, and -9) and three members of the PI3K family (PI3K p110-delta subunit (PIK3CD), PI3K p110-beta subunit (PIK3CB), and PI3K p110-gamma subunit (PIK3CG)) were predicted to be targeted by GBM-N019. Additionally, using GBM-N019 as the query molecules, mTOR, NF-κB, and STAT6 appeared as part of the topmost targets . Molecular docking studies revealed that GBM-N019 docked well into the binding cavity of (Figure 2B), mTOR (Figure 2C), and STAT3 (Figure 2D) with binding affinities of −7.6, −8.1, and −6.9 Kcal/mol, respectively. The ligand receptors complex was bound by several hydrogen bonding, alkyl interactions. Furthermore, the complex was stabilized by various hydrophobic contacts and several Van der Waal forces surrounding the ligand backbone (Figure 1B). Interestingly, GBM-N019 bind to CDK6 with the same affinity that palbociclib has for CDK6 (ΔG = −8.1 Kcal/mol); however, the affinities of GBM-N019 for mTOR and STAT3 are lower than that of their respective standard inhibitors; Dactolisib (ΔG = −9.2 Kcal/mol vs. −7.6 Kcal/mol) and SH-4-54 (ΔG = −7.3 Kcal/mol vs. −6.9 Kcal/mol) . Collectively, our computational study revealed that GBM-N019 is a novel drug candidate as an inhibitor of mTOR/STAT3/CDK6 signature.
Figure 2. Blood–brain barrier permeation and molecular docking profiles of GBM-0N19 with STAT3, mTOR, and CDK6. (A) BBB permeation curve of GBM-N019 as measured by the support vector machine (SVM) and LiCABEDS algorithms of BBB prediction server. The two-dimensional (2D) representations of the ligand–receptor complexes, showing the interacting amino acid residues and the type of interactions occurring between the GBM-N019 and (B) CDK6 (C) mTOR and (D) STAT3.
3.3. GBM-N019 Curbed the Viability and Tumorigenic Features of GBM Cells via Downregulation of NF-κB/Akt/mTOR, STAT3, and CDK6 Signaling In Vitro
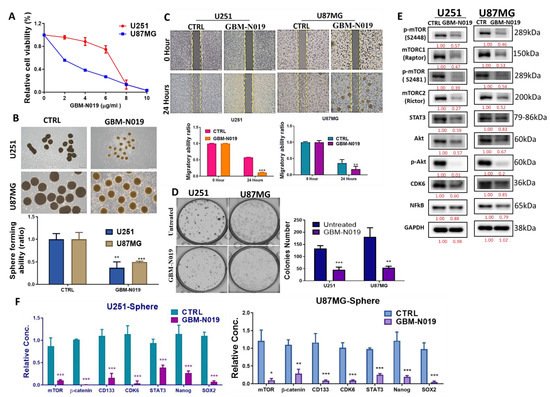
Following our in silico predictions, we evaluated the anti-GBM effects of GBM-N019 using human GBM U251 and U87MG cell lines. GBM-N019 significantly reduced the viability of U251 and U87MG cells in a dose-dependent manner (Figure 3A). Treatment with GBM-N019 also significantly compromised the sphere forming ability (Figure 3B), migratory capacity (Figure 3C), and clonogenicity (Figure 3D) of U251 and U87MG cells. Furthermore, the expression of p-mTOR, mTORC1, mTORC2, STAT3, Akt, p-Akt, CDK6, and NF-κB, in U251 and U87MG cells were significantly downregulated by GBM-N019 treatment (Figure 3E). Our qPCR analysis revealed that the inhibition of the sphere forming ability of U251 and U87MG cells by GBM-N019 was concomitantly associated with decreased expressions of mTOR, β-catenin, CD133, CDK6, STAT3, Nanog, and SOX2 (Figure 3F). Altogether, these findings suggest that GBM-N019 suppressed tumorigenic features of GBM cells via downregulation of NF-κB/Akt/mTOR, STAT3, and CDK6 signaling axis.
Figure 3. GBM-N019 treatment inhibited tumorigenic properties via downregulating mTOR, STAT3, and CDK6 signaling pathways in vitro (A) GBM-N019 treatment significantly hampered the viability of U251 and U87MG cells in dose-dependent manners. GBM-N019 treatment significantly reduced the tumorsphere forming abilities (B), migratory ability (C), and colony forming (D) abilities of both U251 and U87MG cells. (E) Western blots of GBM-N019-treated U251 and U87MG cells exhibited lower expression levels of -mTOR, mTORC1, mTORC2, STAT3, Akt, p-Akt, CDK6, and NF-κB. (F) qPCR analysis showed decreased gene expression levels of mTOR, β-catenin, CD133, CDK6, STAT3, Nanog, and SOX2 in both U251 and U87MG cells after treatment with GBM-N019. * p < 0.05, ** p < 0.01, *** p < 0.001.
3.4. Tumorsphere-Derived Exosomal Cargo of Oncogenes Mediated Treatment Resistance and Aggressive Phenotypes of GBM
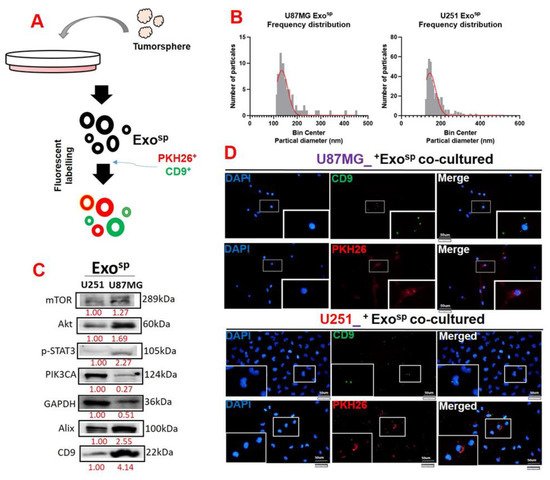
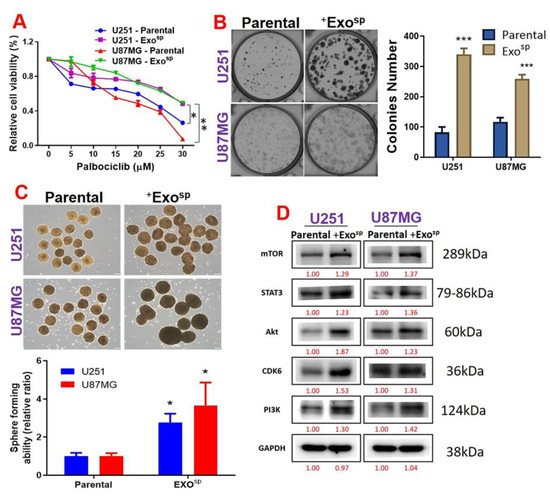
To investigate the oncogenic properties of exosomes, we isolated exosomes from sphere populations of U251 and U87MG cells and cultured parental cells with their respective exosomes. In addition, the isolated exosome (Exosp) contained Akt, mTOR, PIK3CA, and activated STAT3 (p-STAT 3) (Figure 4C). Our immunofluorescence imaging of the U87MG and U251 co-cultured with the isolated Exosp demonstrated that the CD9/PHK26 fluorescent-labeled Exosp were uptaken by the recipient U251 and U87MG cells (Figure 4D). Furthermore, U251 and U87MG cells cultured with the Exosp (ExospU251 and ExospU87MG cells) were less sensitive to palbociclib treatment (Figure 4A) and exhibited higher colony forming (Figure 5B) and sphere generating (Figure 5C) abilities than their respective parental counterparts. Accompanying these tumorigenic features were increased expressions of mTOR, PI3K, Akt, STAT 3, and CDK6 in ExospU251 and ExospU87MG cells compared to the parental cells (Figure 5D).
Figure 4. Characterization of tumorsphere-derived exosomes (Exosp). (A) Schematic representation of exosome isolation from the U87MG and U251 tumorsphere. (B) Particle analysis demonstrated that exosomes (Exosp) isolated from U87MG and U251 tumorspheres were in the 130 and 170 nm diameter range. (C) Western blots of isolated Exosp from U87MG and U251 cells indicated the exosomal cargo of p-STAT3, Akt, mTOR, and PIK3CA. (D) Immunofluorescence imaging depicting the uptake of Exosp in the recipient cells; CD9 (green fluorescence) and PKH26 (red fluorescence) were both exosomal trackers. Numbers in red indicate the relative expression ratio.
Figure 5. Tumorsphere-derived exosomes (Exosp) promoted the expression of mTOR, STAT3 and CDK6 expressions, palbociclib resistance, and tumorigenic properties of GBM cell lines. (A) Cell viability curve showing that U87MG and U251 cells co-cultured with Exosp exhibited low sensitivity to palbociclib treatments compared to their parental counterparts. U87MG and U251 cells co-cultured with Exosp also exhibited higher (B) colony forming ability, (C) sphere generating, and (D) expression levels of mTOR, STAT3, CDK6, Akt, and PI3K compared to their parental counterparts. Numbers in red indicate the relative expression ratio. * p < 0.05, ** p < 0.01, *** p < 0.001.
5. Conclusions
In conclusion, exosomes derived from GSCs mediated the transfer of stemness and invasive properties to U251 and U87MG cells. GBM-N019 efficiently impaired the exchange of oncogenic molecules and rescued cells from the exosomal-mediated gain of drug resistance, stemness, and aggressive properties. Furthermore, GBM-N019 was synergized to enhance the anti-GBM activities of palbociclib. These findings suggested that GBM-N019 possesses good translational relevance as a potential anti-glioblastoma drug candidate worthy of consideration for clinical trials against recurrent glioblastomas.
This entry is adapted from the peer-reviewed paper 10.3390/cells10092391