We explored human glioma single-cell omics studies to find evidence for the assumption that distinct alterations in the genetic background dictate a specific immune TME and influence clinical outcomes. Malignant gliomas display considerable genetic and epigenetic heterogeneity, which affects patient survival and responses to therapy.
Combination of histology with molecular genotyping of key markers: IDH, ATP-dependent helicase (ATRX), Lys-27-Met mutations in histone 3 (H3K27M), TP53 mutations, and 1p/19q chromosomal deletion improved the classification of gliomas and provided some predictors of patient clinical outcome
[4][72]. The transcriptional proneural, classical, and mesenchymal GBM subtypes show varied contribution of tumor-associated glial and microglial cells, and these subtypes associate with a specific TME
[52]. Stratification of WHO grade II-IV glioma patients according to mutational burden indicates that a high number of mutations is associated with significantly worse overall survival and higher expression of immune checkpoint encoding genes
[73][74]. Importantly, patients with high mutational burden showed the enrichment of immune cell signatures
[75].
All these observations suggest a link between a genetic background of the tumor, immune cell composition, and potential responses to immunotherapies. In recent years several single-cell studies have helped to dissect the immune microenvironment of human gliomas and provided evidence for emerging associations between specific tumor somatic mutations and the composition of immune cells within glioma TME.
3.3.1. Impact of IDH Status on the Immune Microenvironment of Gliomas
The
IDH mutation status emerges as a potent modulator of the infiltration of immune cells in glioma TME. Friebel et al. (2020) used a mass cytometry analysis measuring 74 parameters to delineate the myeloid and lymphoid compartment of the brain tumor microenvironment
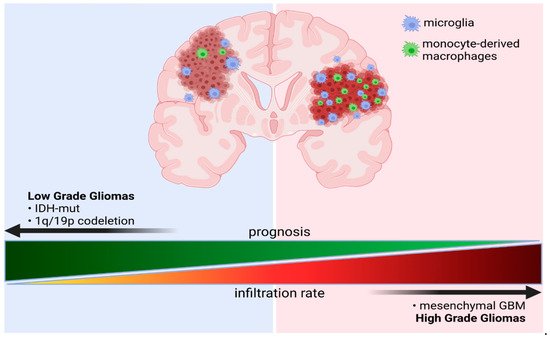
[55]. They showed that GAMs constitute a major immune cell population encompassing up to 80% of all immune cells in the TME. In IDH-wt gliomas monocytes-derived macrophages (MDMs) comprise around 30% of GAMs, whereas in IDH-mut gliomas such cells occur at a very low number and microglial cells dominate the GAM population (
Figure 1). These results were corroborated in a bulk RNA-seq study, in which microglia (CD49d
low) and macrophages (CD49d
high) were separated with FACS sorting
[64]. FACS analysis also confirmed the exclusive expression of CD49d on CNS-invading myeloid cells.
Figure 1. Schematic representation of a ratio of microglia and monocyte-derived macrophages (MDMs) in gliomas of different grades and with distinct genetic backgrounds. Created with BioRender.
Exhaustion, characterized in part by the upregulation of immune checkpoints, contributes to the T-cell dysfunction in GBMs. Levels of inhibitory receptors PD-1, LAG-3/CD223 (lymphocyte activation gene 3 product), TIGIT (T cell immunoreceptor with immunoglobulin and ITIM domain), and ectonucleotidase CD39, combined with T-cell hyporesponsiveness of tumor-specific T cells, suggest poor function of tumor infiltrating lymphocytes (TILs) and confirm severe exhaustion observed in GBM
[76]. These observations have been corroborated by the analyses of CD45+ immunosorted cells from human gliomas. Significant increases in myeloid cells in
IDH-mut and
IDH-
wt gliomas, and lymphocytes in
IDH-
wt tumors, have been reported. Multicolor FACS analyses of 14 major immune cell populations across 100 clinical samples followed by RNA-seq of samples from 48 patients demonstrated the differential abundance of microglia and BM-derived macrophages in IDH-mut and IDH-wt gliomas
[64]. Spatial analysis of tissue sections showed the enrichment of GAMs in the perivascular niche and a closer proximity of MDMs to the vessels compared with activated microglia. Consistently, examination of the transcriptomic data from the Ivy Glioblastoma Atlas Project demonstrated the enrichment of MDMs in the microvascular compartment, in which CD4+ and CD8+ T cells were found in
IDH WT gliomas
[64].
Application of CyTOF (besides dissection of microglia and BM-derived macrophages) revealed changes in other immune cell populations. Among tumor-infiltrating immune cells, Friebel et al. (2020) identified T cells (CD3
+), B cells (CD19
+HLA-DR
+), NK cells (CD56
+CD16
+), neutrophils (CD66b
+), two subsets of classical DCs: cDC1 (CD141
+CADM1
+) and cDC2 (CD1c
+) plasmacytoid DCs: pDCs (CD123
+), and plasma cells (CD19
+, CD38
high). There was no difference in the overall T cell infiltration rate between IDH variants; however, Tregs were significantly more frequent in IDH-wt gliomas and T-cell frequency positively correlated with pDCs and cDCs frequencies, whereas increased numbers of all those populations were associated with decreased GAM/monocyte frequencies
[55]. The
MGMT promoter methylation status did not change frequencies of major immune populations
[55].
In addition to the reduced number of TILs and macrophages, IDH-mut gliomas show lower PD-L1 expression compared to IDH-wt gliomas
[77][78]. A proportion of lymphocytes (T-cells, B-cells, NK cells) was low in IDH-mut gliomas (~10%), whereas in IDH-wt lymphocytes constituted 25% of all immune infiltrating cells
[64]. Additionally, division of T cells into five functional subsets: naïve, central memory (CM), effector memory (EM), terminally differentiated effector memory (TEMRA), and non-circulating tissue-resident (RM), according to the expression of five discriminatory markers (CD45RA, CD45RO, CCR7, CD127, and CD103)
[79], allowed assessing more subtle differences in the T cell populations. Friebel et al. (2020) reported that the majority of T cells found in GBMs were memory T cells
[55]. CD8 RM and EM T-cells had lower expression of proliferation and activation markers in IDH1
mut compared to IDH1
wt gliomas. In contrast, the IDH status had no effect on expression of co-stimulatory (ICOS, CD27, and CD137) and co-inhibitory receptors (2B4, TIGIT, and PD-1)
[55].
The oncometabolite 2-HG inhibits complement activation, complement-mediated phagocytosis, and migration of activated T-cell, their proliferation, and cytokine secretion
[80]. Reduced PD-L1 expression might be a result of lower infiltration of PD-L1 expressing immune cells, but could emerge from epigenetic silencing of the immune checkpoint genes in glioma cells, due to 2-HG-driven DNA hypermethylation
[81][82]. Studies of mouse syngeneic glioma models indicated the lower expression of cytokines in IDH-mut gliomas that might reduce leukocyte chemotaxis
[78]. Still, the mechanism underlying lower infiltration of GAMs in IDH-mut gliomas remains to be elucidated.
Innate lymphoid cells play in anti-tumor immunity and are regulators of TME
[83]. The analysis of NK cells (CD56
+CD3
-) showed that the two main populations of CD56
int/brightCD16
− and CD56
intCD16
+ correspond to the immature and the cytotoxic NK cells, respectively. The enrichment of immature CD56
int/bright was found in CD16
− NK cells among lymphocytes in the IDH1-wt gliomas, whereas predominantly CD56
int/brightCD16
+ NK cells accumulated in the IDH1-mut gliomas. Splitting of the IDH1-wt glioma cohort according to the
MGMT promoter methylation showed a trend toward a higher proportion of CD56
int/brightCD16
+ NK cells in the unmethylated cases. Frequencies of immature NK cells negatively correlated with overall survival in the IDH1-wt cohort (but not significantly)
[55].
Mathewson et al. (2021) investigated T-cell subtypes and expression programs across IDH variants, specifically in glioblastomas
[58]. ScRNA-seq profiling of T cells from 26 IDH-wt and IDH-mut GBMs revealed the presence of CD8 T cells, CD4 conventional T cells (CD4 Tconv), CD4 Tregs, and cycling T cells. Interestingly, the corticosteroid therapy with dexamethasone was associated with substantially reduced numbers of infiltrating T cells (mean reduction was 4.14- and 7.72-fold for CD3+ and CD4+ T cells, respectively). The overall representation of clusters was similar in IDH-wt and IDH-mut GBMs. CD8 and CD4 T cells expressed an interferon signature, an effector memory signature, or a stress signature. The latter was not an artefact, as expression of these genes was detected by RNA in situ hybridization for glioma-infiltrating CD3E+ T cells
[58]. T cell-specific genes (including cytotoxicity genes
PRF1 (
Perforin 1) and
GZMA (
Granzyme a) were more highly expressed in IDH-wt GBMs than IDH-mut GBMs. The cytotoxicity score of CD8 T cells (
PRF1, GZMB, GZMA, GZMH, NKG7, GNLY) was associated with an increased signature of NK cells (
KLRD1,
FGFBP2,
FCGR3A,
S1PR5,
KLRC1,
KLRC3,
KLRB1,
KLRC2). Several NK cell receptor genes, including
KLRC2 (NKG2C protein),
KLRC3 (NKG2E protein),
KLRC1 (NKG2A protein),
KLRD1 (CD94 protein), and
KLRB1 (CD161 protein) were expressed by CD8 T cells with high cytotoxicity scores. These cells represent effectors that share transcriptional profiles with innate T cells, despite having a diverse TCR repertoire. High cytotoxicity of CD8 T cells correlated with lower expression of the
PDCD1 gene (PD-1 protein) and genes coding for co-inhibitory receptors (CTLA4, HAVCR2, LAG3, and TIGIT). The highest level of
PDCD1 was found in cytotoxic CD4 T cells, whereas
TIGIT was most highly expressed by Tregs. The genes coding for the inhibitory CD161 receptor (
KLRB1) and the activating NKG2C/CD94 receptor (
KLRC2 and
KLRD1) were expressed by a large fraction of CD8 T cells.
KLRB1 was preferentially expressed by CD8 and CD4 Tconv cells in diffuse gliomas; its expression was lower in CD4 Tregs. The authors postulated that expression of NK cell receptors is induced in T cells by inflammatory mediators in the glioma TME. CLEC2D, the ligand for CD161, is a surface molecule expressed by immunosuppressive myeloid cells and malignant cells. Mechanistic in vitro and in vivo studies demonstrated that the CD161 receptor inhibits T cell function, including cytotoxicity and cytokine secretion
[58].
3.3.2. The Effects of Co-Deletion of 1p/19q in IDH Mutant Gliomas
Venteicher et al. (2017) performed scRNA-seq analysis of IDH-mut astrocytomas (IDH-A) characterized by
TP53 and
ATRX mutations, and oligodendrogliomas (IDH-O) characterized by mutations in the
TERT (
Telomerase reverse transcriptase) gene promoter and co-deletion of chromosome arms 1p and 19q (1p/19q)
[13]. The authors confirmed the genetic background of the investigated samples at single-cell level. Most genes with higher expression in cells from IDH-A were encoded in 1p/19q regions that are deleted in IDH-O, whereas TP53 targets were enriched in IDH-O as compared with
TP53 mutated IDH-A. The 1p/19q region contains genes encoding potent immunoregulatory proteins including
CSF1 encoded in a 1p region and
TGFB1 encoded in the 19q region. CSF1 is involved in proliferation, differentiation, and survival of myeloid cells and TGFβ1 is a potent immunosuppressive cytokine. Thus, a lack of these genes might negatively influence accumulation of microglia and macrophages in glioma TME. Consistently with this notion, the estimated relative abundance of microglia/macrophages was higher in IDH-A tumors compared to IDH-O tumors
[13].
A recent CyTOF study of the composition of immune infiltrates in IDH-A and IDH-O tumors showed that IDH-A have increased levels of VEGF and TGFβ that play tumor-supportive roles. However, no difference in the proportion of glioma-associated macrophages was noted
[53]. These observations have been supported by the results of CIBERSORT deconvolution of the TCGA dataset combined with IHC staining of LGG samples, that demonstrated a lower number of “M2-related” markers in gliomas with 1p/19q co-deletion
[59]. Co-deletion 1p/19q is a strong, good prognostic marker. Better survival of patients harboring this alteration might be related with reduced infiltration of GAMs, as deletion of the 1p region that encodes the
CSF1 gene yields similar survival outcome as full 1p/19q co-deletion, whereas the effect of 19q deletion is marginal
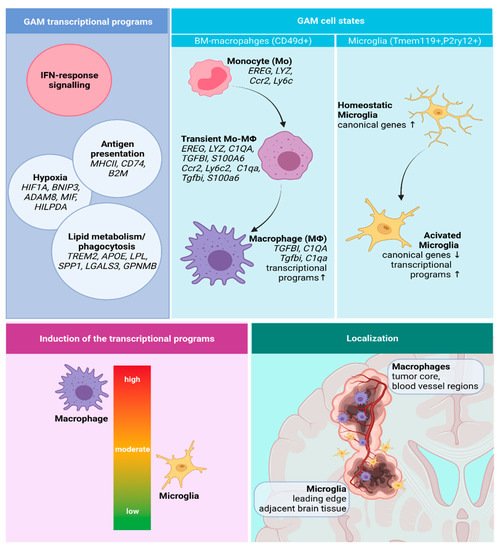
[84]. The identified transcriptional programs of GAMs, their functional states, and localization in GBMs are depicted in the
Figure 2.
Figure 2. A summary of the identified transcriptional programs, cell states, and spatial distribution of GAMs. Created with Biorender.
 +1 credit
+1 credit