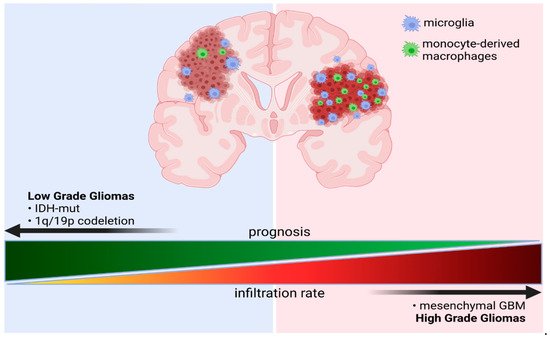
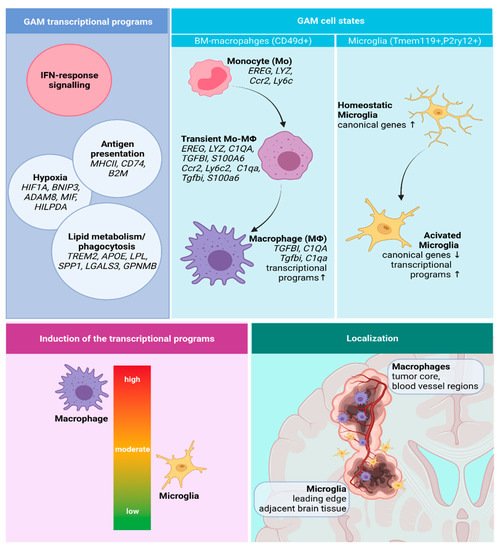
1.1. Classification and Molecular Determinants of Gliomas
Gliomas are tumors of the central nervous system (CNS) that originate from neural stem cells, or astrocytic or oligodendrocytic progenitor cells. Diffuse gliomas represent 80% of primary malignant brain tumors. A new integrated classification system was introduced by the World Health Organization (WHO) in 2016, which comprises five glioma types categorized by both tumor morphology and molecular genetic information
[1]. The largest group contains diffuse gliomas such as astrocytic tumors of the WHO grade II and III, the grade II and III oligodendrogliomas, and the grade IV glioblastomas. A separate category encompasses the WHO grade III and IV diffuse gliomas of childhood. This new classification defines as a separate group lower grade astrocytomas with a more circumscribed growth pattern, lack of
IDH1/2 (isocitrate dehydrogenase 1 coding genes) alterations, and frequent mutations of
BRAF (
v-raf murine sarcoma viral oncogene homolog B1 coding for serine/threonine kinase) (i.e., in pilocytic astrocytoma, pleomorphic xanthoastrocytoma) or
TSC1/
TSC2 (
tuberous sclerosis complex 1/2 encoding hamartin and tuberin) (in subependymal giant cell astrocytoma). Restructuring of the diffuse gliomas classification revealed new subtypes including IDH-wildtype glioblastoma, IDH-mutant glioblastoma, and H3 K27M-mutant diffuse midline glioma
[2].
The
IDH1 gene encodes an isocitrate dehydrogenase 1 which catalyzes the oxidative decarboxylation of isocitrate to α-ketoglutarate (α-KG). Mutations in
IDH1/IDH2 result in the substitution of arginine at codon 132 of the IDH1 or codons 140 or 172 of the IDH2 (IDH1-R132, IDH2-R140 or IDH2-R172). IDH1-R132 mutants have dominant-negative, inhibitory effects on a wild-type IDH1, and gain new functions as they reduce α-KG to its (
R)-enantiomer of 2-hydroxyglutarate (2-HG). Accumulation of 2-HG in cancer cells inhibits Jumonji class histone demethylases and TET (ten-eleven translocation) family of 5-methylcytosine DNA demethylases (reviewed in
[3]). These events result in altered histone methylation and the hypermethylation phenotype
[4]. More than 70% of WHO grade II and III diffuse gliomas are IDH1-R132 mutants.
IDH1/2 mutations in some gliomas are associated with longer survival and better responses to chemotherapy
[5][6][5,6]. Glioblastomas (GBM), divided into an IDH-wildtype (about 90% of cases) and IDH-mutant (about 10% of cases) GBMs, account for 70–75% of all diffuse gliomas and have a median overall survival of 14–17 months. IDH-mutant GBMs are usually secondary glioblastomas with a history of prior lower grade diffuse glioma and are more frequent in younger patients. GBMs encompass also the epithelioid glioblastoma type, giant cell glioblastoma, and gliosarcoma, which are more frequent in children and younger adults, and often harbor a
BRAF V600E mutation.
Comprehensive analyses of genomic transcriptomic alterations in GBMs demonstrated the most common alterations on chromosome 7 (
EGFR/MET/CDK6), chromosome 12 (
CDK4 and
MDM2), and chromosome 4 (
PDGFRA), as well as frequent changes of genes such as
SOX2,
MYCN,
CCND1, and
CCNE2 [7][8][7,8].
EGFR (epidermal growth factor receptor),
PDGFR (platelet derived growth factor receptor), and
MET (MNNG HOS transforming gene) encode receptor tyrosine kinases that control cell proliferation and survival signaling.
NF1 (neurofibromathosis 1),
K-RAS (KIRSTEN rat sarcoma viral oncogene homologue), and
B-RAF are components of signaling pathways controlling proliferation and survival. The cell cycle regulatory protein cyclin D1 (
CCND1), cyclin E2 (
CCNE2), cyclin-dependent kinases 4 and 6 (
CDK4/6), MDM2 (
Mouse double minute 2), and transcription factors SOX2 (
sex-determining region Y-box 2) and MYCN (N-
MYC oncogene) control the cell cycle. Several novel mutations or gene rearrangements were found in genes involved in chromatin organization:
SETD2 (
Set domain containing 2),
ARID2 (
AT-Rich Interaction Domain 2),
DNMT3A (
DNA methyltransferase 3 alpha),
KRAS/NRAS [9]. Aberrant expression or mutations in genes such as
EGFR, NF1, and
PDGFRA/IDH1 allowed the delineation of the classical, mesenchymal, and proneural GBM subtypes, respectively. A tumor transcriptional type and specific genetic alterations confer survival advantages. A weak association between the proneural GBM subtype and longer survival has been reported
[10]. Correlative analyses of transcriptomic profiles showed the survival advantage of the proneural subtype, which is likely to be due to the Glioma CpG island methylator phenotype (G-CIMP). Methylation of the
MGMT (O
6-methylguanine–DNA methyltransferase) gene promoter (resulting in low expression of the DNA repair protein) emerged as a predictive biomarker for treatment response only in a classical GBM subtype
[8][10][8,10]. The subsequent integration of various data sources, multiple methylation, and gene platforms identified new diffuse glioma subgroups in adults. The IDH-mutant non-codel lower grade gliomas (LGG) and GBM were dissected based on the genome-wide patterns of DNA methylation, into two separate subgroups (G-CIMP-low and G-CIMP-high). This dissection is important as the G-CIMP-low subset had an unfavorable clinical outcome
[9].
Introduction of scRNA-seq to GBM studies revealed their considerable genetic heterogeneity and discrete functional states. Implementation of single cell RNA sequencing (scRNA-seq) to glioma studies showed the considerable intratumoral genetic heterogeneity and mosaic expression of genes coding for surface receptors and ligands, including
EGFR,
PDGFRA,
PDGFA,
FGFR1 (
fibroblast growth factor receptor 1),
FGF1 (
fibroblast growth factor 1),
NOTCH2 (
notch receptor 2), and
JAG1 (
jagged canonical notch ligand 1)
[11]. Transcriptomic analyses revealed four meta-signatures related to hypoxia, complement/immune response, oligodendrocytes, and cell cycle. Application of the stemness signature (
POU3F2,
SOX2,
SALL2,
OLIG2) demonstrated the presence of glioma stem cells.
POU3F2 (
POU Class 3 Homeobox 2),
SOX2,
SALL2 (
Spalt Like Transcription Factor 2), and
OLIG2 (
Oligodendrocyte transcription factor 2) encode transcription factors related to the maintenance of the stemness phenotype. Mesenchymal tumors with high expression of stemness markers (
POU3F2,
SOX2,
SALL2,
OLIG2) had significantly worse outcome than the proneural subtype tumors
[11]. Integrative studies of scRNA-seq data of GBMs, combined with genomic and bulk gene expression analyses collected in The Cancer Genome Atlas (TCGA), allowed the definition of discrete functional GBM states. Copy number amplifications of the
CDK4,
EGFR, and
PDGFRA loci and mutations in the
NF1 locus define GBM states: neural progenitor-like (NPC-like), oligodendrocyte-progenitor-like (OPC-like), astrocyte-like (AC-like), and mesenchymal-like (MES-like) states. While their relative frequency varies between tumors, genetic alterations in
CDK4,
PDGFRA,
EGFR, and
NF1 define a predominant state
[12][13][12,13].
EGFR aberrations were associated with a predominance of the AC-like cells, while amplifications of
CDK4 and
PDGFRA were connected with the prevalence the NPC-like and OPC-like states, respectively. Chr5q deletions and
NF1 alterations affected the frequency of MES-like states
[12]. Collective data suggest that the molecularly distinct GBM subtypes representing various functional states have a profound impact on their microenvironment and this notion is supported by the data from mouse GBM models showing that well-defined driver mutations may create unique microenvironments
[14].