 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Jiun-Yang Chiang | + 1938 word(s) | 1938 | 2021-08-26 10:20:41 | | | |
| 2 | Vicky Zhou | Meta information modification | 1938 | 2021-09-07 06:47:29 | | |
Video Upload Options
Very low-density lipoprotein (VLDL) and chylomicrons, which are known as TG-rich lipoproteins (TRLs), are spherical particles with core lipids (TG and cholesterol esters), phospholipids, free cholesterol, and surface apolipoproteins. The origins of TGs are generally exogenous or endogenous. Exogenous TG is mostly obtained from daily diet and transported within chylomicrons, while endogenous TG circulates in VLDL and is mostly formed in the hepatobiliary system.
1. Introduction
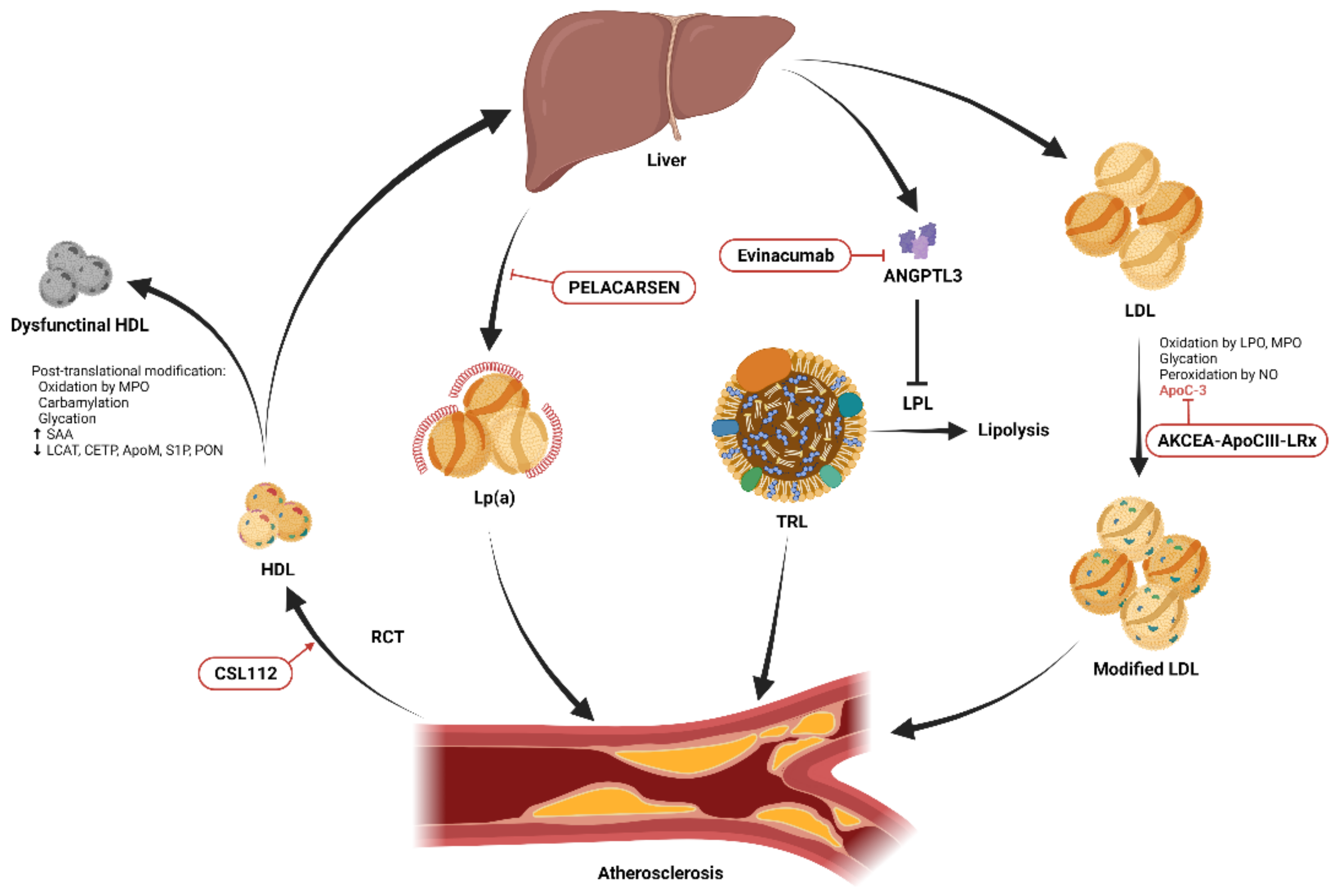
Figure 1. Schematic diagram showing the essential modifications of low-density lipoprotein (LDL) and high-density lipoprotein (HDL) and potential therapeutic targets. HDL promotes cholesterol efflux from cells within atherosclerotic plaques through reverse cholesterol transport (RCT) and transports excess cholesterol from peripheral tissues to the liver for excretion. Post-translational modifications including oxidation, carbamylation, glycation, and alterations of its lipidomic and proteomic structure result in dysfunctional HDL. Infusion of reconstituted apolipoprotein A-I, CSL112, enhances RCT. Lipoprotein(a) (Lp(a)) promotes atherosclerosis through its proinflammatory and antifibrinolytic effects. The production of Lp(a) in the liver can be reduced by the novel antisense oligonucleotide, PELACARSEN. Angiopoietin-like protein 3 (ANGPTL3) produced in the liver inhibits lipoprotein lipase (LPL)-induced lipolysis, resulting in increased circulating triglycerides carried by triglyceride-rich lipoproteins (TRLs), and accelerated atherosclerosis [9]. This process can be blocked by the ANGPTL3 inhibitor, evinacumab. Native LDL is modified by oxidation, glycation, peroxidation, and apolipoprotein C-III (apoC-III) adhesion and becomes more atherogenic. The expression of apoC-III can be suppressed by another novel antisense oligonucleotide, AKCEA-ApoCIII-LRx.
2. Triglycerides
| Potential Therapeutic Target | Pharmacological Approach | Published Clinical Trials | Subjects | Pros | Cons | Ongoing Trials and The Aims of The Trials |
|---|---|---|---|---|---|---|
| Angiopoietin-like protein 3 | Evinacumab, a recombinant human monoclonal antibody that inhibits angiopoietin-like protein 3 | ELIPSE HoFH (phase 3) [32] | Patients with homozygous familial hypercholesterolemia |
|
|
|
| ApoC-III | Volanesorsen, a 2ʹ-O-(2-methoxyethyl)-modified antisense oligonucleotide | APPROACH (phase 3) [34] | Patients with familial chylomicronemia syndrome |
|
|
|
| COMPASS (phase 3) [35] | Patients with severe hypertriglyceridemia |
|
||||
| AKCEA-ApoCIII-LRx, a GalNAc3 modified antisense oligonucleotide | Phase 1/2a trial [36] | Healthy volunteers |
|
|
||
| ApoA-I | CSL112, a plasma-derived reconstituted apoA-I | Phase 2b AEGIS-I trial [37] | Patients with myocardial infarction |
|
|
|
| Apolipoprotein(a) | PELACARSEN, an GalNAC3 modified antisense oligonucleotide | Phase 2 trial [38] | Patients with established ASCVD |
|
|
3. Conclusions
References
- Mensah, G.A.; Wei, G.S.; Sorlie, P.D.; Fine, L.J.; Rosenberg, Y.; Kaufmann, P.G.; Mussolino, M.E.; Hsu, L.; Addou, E.; Engelgau, M.M.; et al. Decline in Cardiovascular Mortality: Possible Causes and Implications. Circ. Res. 2017, 120, 366–380.
- Amini, M.; Zayeri, F.; Salehi, M. Trend analysis of cardiovascular disease mortality, incidence, and mortality-to-incidence ratio: Results from global burden of disease study 2017. BMC Public Health 2021, 21, 1–12.
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Braun, L.T.; De Ferranti, S.; Faiella-Tommasino, J.; Forman, D.E.; et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guideline. Circulation 2019, 139, e1082–e1143.
- Banach, M.; Duell, P.B.; Gotto, A.M., Jr.; Laufs, U.; Leiter, L.A.; Mancini, G.B.J.; Ray, K.K.; Flaim, J.; Ye, Z.; Catapano, A.L. Association of Bempedoic Acid Administration with Atherogenic Lipid Levels in Phase 3 Randomized Clinical Trials of Patients with Hypercholesterolemia. JAMA Cardiol. 2020, 5, 1124–1135.
- Cicero, A.F.G.; Pontremoli, R.; Fogacci, F.; Viazzi, F.; Borghi, C. Effect of Bempedoic Acid on Serum Uric Acid and Related Outcomes: A Systematic Review and Meta-analysis of the available Phase 2 and Phase 3 Clinical Studies. Drug Saf. 2020, 43, 727–736.
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131.
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188.
- Yoshida, H.; Kisugi, R. Mechanisms of LDL oxidation. Clin. Chim. Acta 2010, 411, 1875–1882.
- Ruscica, M.; Macchi, C.; Fogacci, F.; Ferri, N.; Grandi, E.; Rizzoli, E.; D’Addato, S.; Borghi, C.; Cicero, A.F. Angiopoietin-like 3 and subclinical peripheral arterial disease: Evidence from the Brisighella Heart Study. Eur. J. Prev. Cardiol. 2020, 27, 2251–2254.
- Dron, J.S.; Wang, J.; Cao, H.; McIntyre, A.D.; Iacocca, M.A.; Menard, J.R.; Movsesyan, I.; Malloy, M.J.; Pullinger, C.R.; Kane, J.P.; et al. Severe hypertriglyceridemia is primarily polygenic. J. Clin. Lipidol. 2019, 13, 80–88.
- Truthmann, J.; Schienkiewitz, A.; Busch, M.A.; Mensink, G.B.M.; Du, Y.; Bosy-Westphal, A.; Knopf, H.; Scheidt-Nave, C. Changes in mean serum lipids among adults in Germany: Results from National Health Surveys 1997-99 and 2008-11. BMC Public Health 2016, 16, 240.
- Hegele, R.A.; Ginsberg, H.N.; Chapman, M.J.; Nordestgaard, B.G.; Kuivenhoven, J.A.; Averna, M.; Borén, J.; Bruckert, E.; Catapano, A.L.; Descamps, O.S.; et al. The polygenic nature of hypertriglyceridaemia: Implications for definition, diagnosis, and management. Lancet Diabetes Endocrinol. 2014, 2, 655–666.
- Xiao, C.; Dash, S.; Morgantini, C.; Hegele, R.A.; Lewis, G.F. Pharmacological Targeting of the Atherogenic Dyslipidemia Complex: The Next Frontier in CVD Prevention Beyond Lowering LDL Cholesterol. Diabetes 2016, 65, 1767–1778.
- Stahel, P.; Xiao, C.; Hegele, R.A.; Lewis, G.F. The Atherogenic Dyslipidemia Complex and Novel Approaches to Cardiovascular Disease Prevention in Diabetes. Can. J. Cardiol. 2018, 34, 595–604.
- Ganda, O.P.; Bhatt, D.L.; Mason, R.P.; Miller, M.; Boden, W.E. Unmet Need for Adjunctive Dyslipidemia Therapy in Hypertriglyceridemia Management. J. Am. Coll. Cardiol. 2018, 72, 330–343.
- Nordestgaard, B.G.; Varbo, A. Triglycerides and cardiovascular disease. Lancet 2014, 384, 626–635.
- Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Unmet need for primary prevention in individuals with hypertriglyceridaemia not eligible for statin therapy according to European Society of Cardiology/European Atherosclerosis Society guidelines: A contemporary population-based study. Eur. Heart J. 2018, 39, 610–619.
- Schwartz, G.G.; Abt, M.; Bao, W.; DeMicco, D.; Kallend, D.; Miller, M.; Mundl, H.; Olsson, A.G. Fasting Triglycerides Predict Recurrent Ischemic Events in Patients with Acute Coronary Syndrome Treated with Statins. J. Am. Coll. Cardiol. 2015, 65, 2267–2275.
- Sarwar, N.; Sandhu, M.S.; Ricketts, S.L.; Butterworth, A.S.; Di Angelantonio, E.; Boekholdt, M.; Ouwehand, W.; Watkins, H.; Samani, N.J.; Saleheen, D.; et al. Triglyceride-mediated pathways and coronary disease: Collaborative analysis of 101 studies. Lancet 2010, 375, 1634–1639.
- Dron, J.S.; Hegele, R.A. Complexity of mechanisms among human proprotein convertase subtilisin-kexin type 9 variants. Curr. Opin. Lipidol. 2017, 28, 161–169.
- Ference, B.A.; Kastelein, J.J.P.; Ray, K.K.; Ginsberg, H.N.; Chapman, M.J.; Packard, C.J.; Laufs, U.; Oliver-Williams, C.; Wood, A.M.; Butterworth, A.S.; et al. Association of Triglyceride-Lowering LPL Variants and LDL-C–Lowering LDLR Variants with Risk of Coronary Heart Disease. JAMA 2019, 321, 364–373.
- Sarwar, N.; Danesh, J.; Eiriksdottir, G.; Sigurdsson, G.; Wareham, N.; Bingham, S.; Boekholdt, S.M.; Khaw, K.-T.; Gudnason, V. Triglycerides and the Risk of Coronary Heart Disease. Circulation 2007, 115, 450–458.
- Di Angelantonio, E.; Gao, P.; Pennells, L.; Kaptoge, S.; Caslake, M.J.; Thompson, A.; Butterworth, A.S.; Sarwar, N.; Wormser, D.; Saleheen, D.; et al. Lipid-Related Markers and Cardiovascular Disease Prediction. JAMA 2012, 307, 2499–2506.
- Laufs, U.; Parhofer, K.G.; Ginsberg, H.N.; Hegele, R.A. Clinical review on triglycerides. Eur. Heart J. 2020, 41, 99–109c.
- Willer, C.J.; Schmidt, E.M.; Sengupta, S.; Peloso, G.M.; Gustafsson, S.; Kanoni, S.; Ganna, A.; Chen, J.; Buchkovich, M.L.; Mora, S.; et al. Discovery and refinement of loci associated with lipid levels: A Systematic Review and Meta-analysis. Nat. Genet. 2013, 45, 1274–1283.
- Silverman, M.G.; Ference, B.A.; Im, K.; Wiviott, S.D.; Giugliano, R.P.; Grundy, S.M.; Braunwald, E.; Sabatine, M.S. Association Between Lowering LDL-C and Cardiovascular Risk Reduction Among Different Therapeutic Interventions. JAMA 2016, 316, 1289–1297.
- Ference, B.A.; Yoo, W.; Alesh, I.; Mahajan, N.; Mirowska, K.K.; Mewada, A.; Kahn, J.; Afonso, L.; Williams, K.A.; Flack, J.M. Effect of Long-Term Exposure to Lower Low-Density Lipoprotein Cholesterol Beginning Early in Life on the Risk of Coronary Heart Disease: A Mendelian Randomization Analysis. J. Am. Coll. Cardiol. 2012, 60, 2631–2639.
- Holmes, M.V.; Asselbergs, F.W.; Palmer, T.M.; Drenos, F.; Lanktree, M.B.; Nelson, C.P.; Dale, C.E.; Padmanabhan, S.; Finan, C.; Swerdlow, D.I.; et al. Mendelian randomization of blood lipids for coronary heart disease. Eur. Heart J. 2015, 36, 539–550.
- Razi, F.; Forouzanfar, K.; Bandarian, F.; Nasli-Esfahani, E. LDL-cholesterol measurement in diabetic type 2 patients: A comparison between direct assay and popular equations. J. Diabetes Metab. Disord. 2017, 16, 43.
- Langlois, M.R.; Descamps, O.S.; van der Laarse, A.; Weykamp, C.; Baum, H.; Pulkki, K.; von Eckardstein, A.; De Bacquer, D.; Borén, J.; Wiklund, O.; et al. Clinical impact of direct HDLc and LDLc method bias in hypertriglyceridemia. A simulation study of the EAS-EFLM Collaborative Project Group. Atherosclerosis 2014, 233, 83–90.
- Baca, A.M.; Warnick, G.R. Estimation of LDL-Associated Apolipoprotein B from Measurements of Triglycerides and Total Apolipoprotein B. Clin. Chem. 2008, 54, 907–910.
- Raal, F.J.; Rosenson, R.S.; Reeskamp, L.F.; Hovingh, G.K.; Kastelein, J.J.P.; Rubba, P.; Ali, S.; Banerjee, P.; Chan, K.-C.; Gipe, D.A.; et al. Evinacumab for Homozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 383, 711–720.
- Kuehn, B.M. Evinacumab Approval Adds a New Option for Homozygous Familial Hypercholesterolemia with a Hefty Price Tag. Circulation 2021, 143, 2494–2496.
- Witztum, J.L.; Gaudet, D.; Freedman, S.D.; Alexander, V.J.; Digenio, A.; Williams, K.R.; Yang, Q.; Hughes, S.G.; Geary, R.S.; Arca, M.; et al. Volanesorsen and Triglyceride Levels in Familial Chylomicronemia Syndrome. N. Engl. J. Med. 2019, 381, 531–542.
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’Dea, L.S.L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275.
- Alexander, V.J.; Xia, S.; Hurh, E.; Hughes, S.G.; O’Dea, L.; Geary, R.S.; Witztum, J.L.; Tsimikas, S. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur. Heart J. 2019, 40, 2785–2796.
- Michael Gibson, C.; Korjian, S.; Tricoci, P.; Daaboul, Y.; Yee, M.; Jain, P.; Alexander, J.H.; Steg, P.G.; Lincoff, A.M.; Kastelein, J.J.; et al. Safety and Tolerability of CSL112, a Reconstituted, Infusible, Plasma-Derived Apolipoprotein A-I, After Acute Myocardial Infarction: The AEGIS-I Trial (ApoA-I Event Reducing in Ischemic Syndromes I). Circulation 2016, 134, 1918–1930.
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255.

