 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Gino Seravalle | + 1308 word(s) | 1308 | 2021-08-18 08:48:00 | | | |
| 2 | Vicky Zhou | Meta information modification | 1308 | 2021-09-03 08:11:56 | | |
Video Upload Options
Sympathetic–vascular interactions means that the sympathetic nervous system plays a pivotal role in the short- and long-term regulation of different cardiovascular functions. In recent decades, increasing evidence has demonstrated that sympathetic neural influences are involved not only in the vasomotor modulation of small resistance arteries but also in the control of large arteries. Sympathetic activity and vascular function, which are key factors in the pathophysiology and prognosis of cardiovascular disease, are linked by a close relationship. Evidence from experimental studies indicates that the sympathetic nervous system is critically influenced, at the central and also at the peripheral level, by the most relevant factors regulating vascular function, namely nitric oxide, reactive oxygen species and endothelin. Additionally, there is evidence of a reciprocal influence between endothelial function and sympathetic mechanisms.
1. Introduction
Basic and clinical studies carried out during recent decades have allowed to substantially improve the available information on the role of the sympathetic nervous system in the regulation of cardiovascular function in physiological conditions and in pathologic states directly or indirectly affecting the heart and systemic circulation [1]. Evidence has been provided that sympathetic neural influences, in conjunction with other circulating humoral factors, exert a fundamental role in the homeostatic control of the cardiovascular system [1]. Such interactions represent the key pathophysiological steps in the development and progression of several cardiovascular diseases, becoming important triggers of complications, markers of prognosis and targets of treatment.
An increase in sympathetic activity may reduce arterial distensibility via at least two non-mutually exclusive mechanisms. First, distensibility may be reduced because the resulting increase in vessel diameter stretches the less distensible component of the vessel wall (e.g., collagen), making the relationship an inverse one [2][3][4]. Second, distensibility can be reduced because of a sympathetic-dependent increase in heart rate, given that this increase is associated with a stiffening of medium-sized and large elastic arteries in both animals and humans [5][6]. An additional mechanism may consist in contraction of the vascular smooth muscle because the elastic modulus of contracted muscle tissue is greater than that of the relaxed one [7].
2. Sympathetic–Vascular Alterations in Cardiovascular Disease
The vast majority of clinical conditions known to be characterized by an adrenergic overdrive, including essential hypertension, congestive heart failure, chronic kidney disease, obesity and metabolic syndrome, share endothelial dysfunction as a common hallmark [7]. It should be emphasized that in some of the above conditions, sympathetic overdrive and, concomitantly, endothelial dysfunction have a clear-cut clinical relevance because both have been shown to represent independent markers of the disease prognosis [8][9][10][11][12].
The increased sympathetic activity reported in clinical conditions characterized by high blood pressure appears to be a peculiarity of the essential hypertensive state, being undetectable in the secondary forms of the disease [13]. Furthermore, various studies have convincingly shown that the magnitude of the adrenergic overdrive parallels the severity of the hypertensive state, becoming more and more elevated as the clinic or ambulatory blood pressure values progressively increase [13]. Although several studies support the “central neurogenic “ nature of the hypertension-related sympathetic overdrive, evidence has been provided showing that a decrease in nitric oxide levels can “per se” at least in part be responsible for the augmented sympathetic neural drive [14][15]. Similarly, in patients with chronic kidney disease, also characterized by a marked sympathetic activation [9], circulating plasma levels of asymmetric dimethylarginine, an inhibitor of nitric oxide, are elevated [16].
The situation appears to be more complex in congestive heart failure, which is characterized by functional and structural cardiovascular alterations which involve the cardiac pump and the peripheral circulation as well. The latter include, among others, a reduction in arterial distensibility and, thus, an increase in arterial stiffness, which may favor the progression of the heart failure state by determining changes in central hemodynamics [17]. A number of factors, alone or in combination, participate in the development and progression of the arterial stiffness alterations, including sympathetic activation [18]. It is worth mentioning that, firstly, a significant relationship exists between the reduction in arterial distensibility (and thus the increase in arterial stiffness) and the magnitude of the adrenergic overdrive, and secondly, cardiac sympathetic activation appears to be closely linked to pulmonary arterial pressure, independently of heart failure etiology [19]. Regarding the mechanisms responsible for the neuroadrenergic overdrive characterizing heart failure, growing evidence supports the notion that the central sympathoinhibitory effects exerted by nitric oxide appear to be impaired in this condition, presumably because of a decrease in the availability of neuronal nitric oxide synthase [20]. This represents the pathophysiological background for the finding that endothelial dysfunction appears to be a hallmark of the heart failure state. In this context, it is worth mentioning that both endothelial (and more generally vascular) alterations and adrenergic overdrive share a common behavior. Namely, the alterations occur early in the clinical course of the disease, being detectable in a mild heart failure state (patients belonging to New York Heart Association functional classes I and II) and becoming more and more pronounced as the clinical severity of the disease increases ( Figure 1 ) [21].
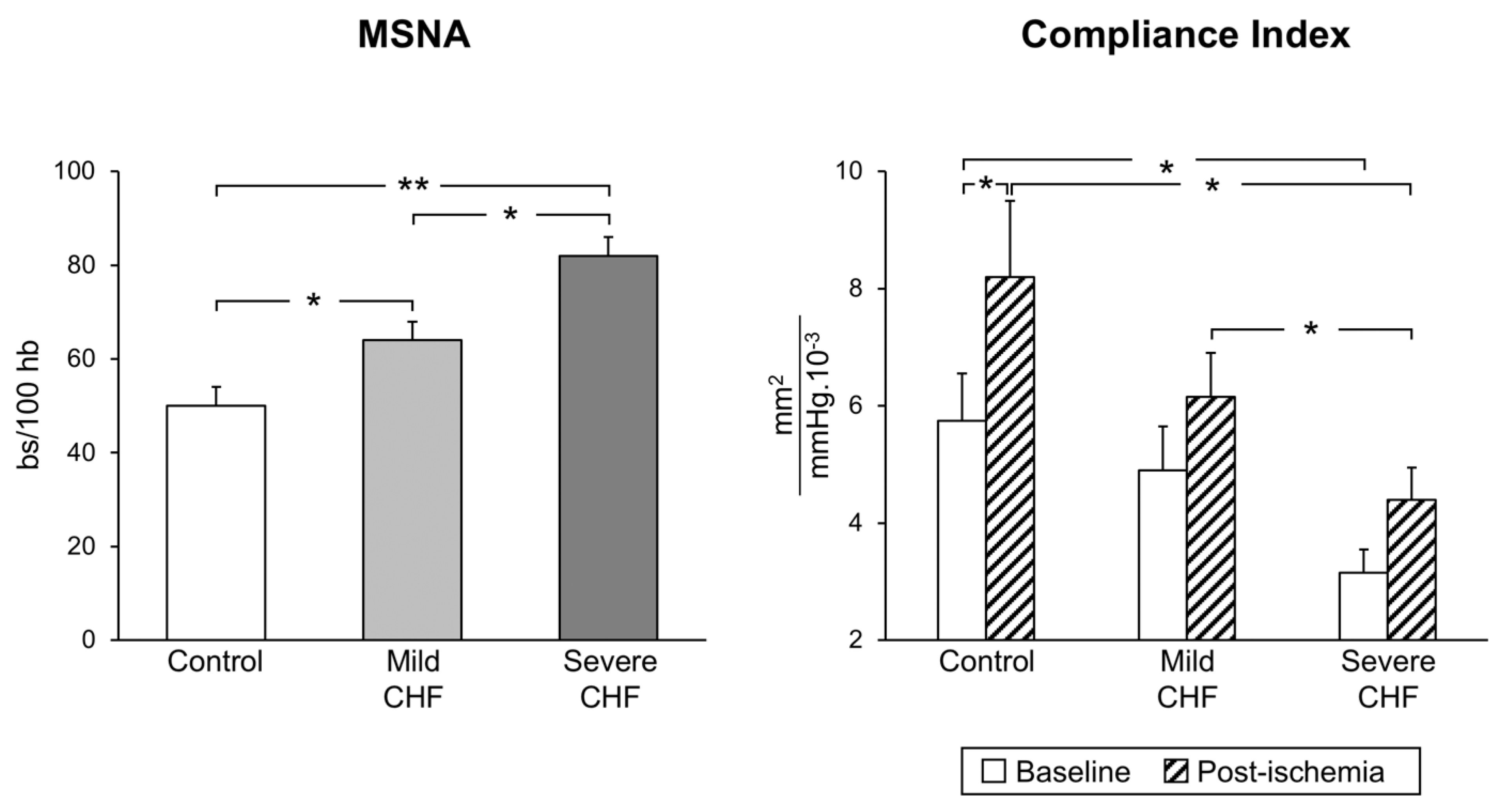
Figure 1. Progressive increase in sympathetic nerve traffic (left panel, MSNA, as assessed via microneurography) and progressive decrease in arterial distensibility (compliance index, right panel) from the healthy state (control subjects) to patients with mild and severe congestive heart failure (CHF). Bs: bursts; HB: heart beats. Asterisks (* p < 0.05, ** p < 0.01) refer to the statistical significance between groups. From data presented in Ref. [21].
An intriguing question is whether and to what extent the sympathetic abnormalities described in hypertension and in congestive heart failure can be reversed by drug treatment. Evidence has been provided that different pharmacological interventions may favor a partial restoration (although not a full normalization) of the sympathetic abnormalities described in the abovementioned clinical conditions [18][22]. This may particularly be the case for drugs, such as angiotensin-converting enzyme inhibitors, angiotensin II receptor blockers and, among beta blockers, nebivolol, which combine the sympathomoderating effects with a favorable action on nitric oxide.
Cardiometabolic diseases characterized by a marked sympathetic activation, such as obesity and metabolic syndrome [23], also display profound alterations in the structural and functional characteristics of large and medium arteries. These abnormalities, which include an increase in arterial stiffness, a reduction in arterial distensibility and an impairment of endothelial function, have also been reported in small resistance arteries [23][24]. Given the close relationship found between the functional and structural alterations described in the coronary and subcutaneous small arteries, these vascular and endothelial abnormalities may have important implications for the progression of organ damage related to cardiovascular risk factors and for determining cardiovascular prognosis [25].
3. Concluding Remarks
References
- Mancia, G.; Luscher, T.F.; Shepherd, J.T.; Noll, G.; Grassi, G. Cardiovascular regulation: Basic considerations. In Cardiovascular Medicine, 3rd ed.; Willerson, J.T., Cohn, J.N., Wellens, H.J.J., Holmes, D., Eds.; Springer: London, UK, 2007; pp. 1525–1540.
- Lacolley, P.; Ragnault, V.; Laurent, S. Mechanisms of arterial stiffening. Ather. Thromb. Vasc. Biol. 2020, 40, 1055–1062.
- Mäki-Petäjä, K.M.; Barrett, S.M.; Evans, S.V.; Cheriyan, J.; McEniery, C.M.; Wilkinson, I.B. The role of the autonomic nervous system in the regulation of aortic stiffness. Hypertension 2016, 68, 1290–1297.
- Nardone, M.; Floras, J.; Millar, P.J. Sympathetic neural modulation of arterial stiffness in humans. Am. J. Physiol. 2020, 319, H1338–H1346.
- Grassi, G. Impact of heart rate on arterial stiffness: Virtual vs real assessment. J. Hypertens. 2020, 38, 2382–2383.
- Mancia, G.; Masi, S.; Palatini, P.; Tsioufis, C.; Grassi, G. Elevated heart rate and cardiovascular risk in hypertension. J. Hypertens. 2021, 39, 1060–1069.
- Seravalle, G.; Grassi, G.; Mancia, G. Arterial alterations in hypertension. In Arterial Disorders; Berbari, A., Mancia, G., Eds.; Springer International Publishing: Basel, Switzerland, 2015; pp. 285–298.
- Cohn, J.; Levine, T.; Olivari, M.T.; Garberg, V.; Tura, D.; Francis, G.S.; Simon, A.B.; Rector, T. Plasma norepinephrine as a guide to prognosis in patients with congestive heart failure. N. Engl. J. Med. 1984, 311, 819–823.
- Zoccali, C.; Mallamaci, F.; Parlongo, S.; Cutrupi, S.; Benedetto, F.C.; Tripepi, G.; Bonanno, G.; Rapisarda, G.F.; Fatuzzo, P.; Seminara, G.; et al. Plasma norepinephrine predicts survival and incident cardiovascular events in patients with end-stage renal disease. Circulation 2002, 105, 1354–1359.
- Halcox, J.P.; Schenke, W.H.; Zalos, G.; Mincemoyer, R.; Prasad, A.; Waclawiw, H.R.A.; Quyyumi, A.A. Prognostic value of coronary vascular endothelial dysfunction. Circulation 2002, 106, 653–658.
- Targonski, P.V.; Bonetti, P.O.; Pumper, G.M.; Higano, S.T.; Holmes, D.R., Jr.; Lerman, A. Coronary endothelial dysfunction is associated with an increased risk of cerebrovascular events. Circulation 2003, 107, 2805–2809.
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368.
- Grassi, G.; Mark, A.; Esler, M. The sympathetic nervous system alterations in human hypertension. Circ. Res. 2015, 116, 976–990.
- Gamboa, A.; Okamoto, L.E.; Diedrich, A.; Choi, L.; Robertson, D.; Farley, G.; Paranjape, S.; Biaggioni, I. Sympathetic activation and nitric oxide function in early hypertension. Am. J. Physiol. 2012, 302, H1438–H1443.
- Quarti-Trevano, F.; Seravalle, G.; Dell’Oro, R.; Mancia, G.; Grassi, G. Autonomic cardiovascular alterations in chronic kidney disease: Effects of dialysis, kidney transplantation and renal denervation. Curr. Hypertens. Rep. 2021, 23, 10.
- Grassi, G.; Seravalle, G.; Ghiadoni, L.; Tripepi, G.; Bruno, R.M.; Mancia, G.; Zoccali, C. Sympathetic nerve traffic and asymmetric dimethylarginine in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 2620–2627.
- Weber, T.; Chirinos, J.A. Pulsatile arterial haemodynamics in heart failure. Eur. Heart. J. 2018, 39, 1–11.
- Grassi, G.; Mancia, G.; Esler, M.D. Central and peripheral sympathetic activation in heart failure. Cardiovasc. Res. 2021, in press.
- Kaye, D.M.; Smirk, B.; Finch, S.; Williams, C.; Esker, M.D. Intercation between cardiac sympathetic drive and heart rate in heart failure: Modulation of adrenergic receptor genotype. J. Am. Coll. Cardiol. 2004, 44, 2008–2015.
- Farah, C.; Michel, L.Y.M.; Balligand, J.L. Nitric oxide signalling in cardiovascular health and disease. Nat. Rev. Cardiol. 2018, 15, 292–316.
- Grassi, G.; Giannattasio, C.; Failla, M.; Pesenti, A.; Peretti, E.; Marinoni, N.; Fraschini, S.; Vailati, S.; Mancia, G. Sympathetic modulation of radial artery compliance in congestive heart failure. Hypertension 1995, 26, 348–354.
- Grassi, G. Sympathomoderating effects of antihypertensive drug treatment. Am. J. Hypertens. 2016, 29, 665–675.
- Grassi, G.; Seravalle, G.; Brambilla, G.; Facchetti, R.; Bolla, G.; Mozzi, E.; Mancia, G. Impact of the metabolic syndrome on subcutaneous microcirculation in obese patients. J. Hypertens. 2010, 28, 1708–1714.
- Grassi, G.; Seravalle, G.; Scopelliti, F.; Dell’Oro, R.; Fattori, L.; Quarti-Trevano, F.; Brambilla, G.; Schiffrin, E.L.; Mancia, G. Structural and functional alterations of subcutaneous small resistance arteries in severe human obesity. Obesity 2010, 18, 92–98.
- Masi, S.; Georgiopoulos, G.; Chiriacò, M.; Grassi, G.; Seravalle, G.; Savoia, C.; Volpe, M.; Taddei, S.; Rizzoni, D. The importance of endothelial dysfunction in resistance artery remodelling and cardiovascular risk. Cardiovasc. Res. 2020, 116, 429–437.
- Monteiro, J.P.; Bennet, M.; Rodor, J.; Caudrillier, A.; Ulitisky, I.; Baker, A.H. Endothelial function and dysfunction in the cardiovascular system: The long non-coding road. Cardiovasc. Res. 2019, 115, 1692–1704.
- Toh, B.H.; Bobik, A.; Kyaw, T.S.; Drummond, G.R.; Sobey, C.G.; Guzik, T.J. Immune mechanisms in vascular disease and stroke. Biomed. Res. 2014, 2014, 730691.
- Dhindsa, D.S.; Sandesara, P.B.; Shapiro, M.D.; Wong, N.D. The evolving understanding and approach to cardiovascular risk management. Front. Cardiovasc. Med. 2020.

