 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Vincent Chagnault | + 1259 word(s) | 1259 | 2021-08-30 10:10:26 | | | |
| 2 | Nora Tang | + 245 word(s) | 1504 | 2021-09-03 03:20:26 | | |
Video Upload Options
The chemical synthesis of linear high DP oligomaltoses (OMs) is much more efficient by the opening of cyclodextrins (CDs). The α, β, and γ-CDs are cyclic oligosaccharides composed of 6, 7, or 8 glucose units respectively linked by a α-1,4 glycosidic bond. They are industrially prepared using CD glucanotransferase on starch.
1. Introduction
While enzymatic catalysis leads to mixtures produced in relatively small amounts, the chemical synthesis of pure linear OM with high DP is complex. For example, the total synthesis of hexamaltooligosaccharides with various protecting groups was achieved by Takahashi et al. in 14 steps from maltose in 4% overall yield [1]. Starting from these products, they pursued their work by synthesizing octamaltooligosaccharides in 21% yield using a protected β-maltosyl fluoride donor [2].
The chemical synthesis of linear high DP oligomaltoses (OMs) is much more efficient by the opening of cyclodextrins (CDs). The α, β, and γ-CDs are cyclic oligosaccharides composed of 6, 7, or 8 glucose units respectively linked by a α-1,4 glycosidic bond ( Scheme 1 ). They are industrially prepared using CD glucanotransferase on starch [3]. Very recently, β-CD was also prepared with 70% yield from maltose using the same enzyme and 1-adamantane carboxylic acid as supramolecular template to control the reaction [4].
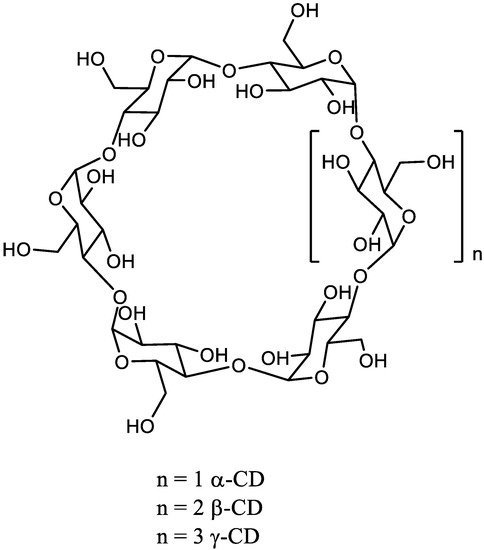
CDs are available in rather large quantity with well-characterized structures. Among CDs, the β-CD is a fairly cheap raw material.
Firstly, we will review herein the enzymatic opening of CDs. Then the different reactions described in literature for OM preparations, by chemical ring-opening of CDs, will be described. Finally, we will discuss the different applications of these molecules of interest through further functionalization.
2. Enzymatic Ring-Opening of Cyclodextrins
A review of CD degrading enzymes (Cyclomaltodextrinases, EC 3.2.1.54, cyclomaltodextrin hydrolases) and α-amylases from microbial sources ( Bacillus coagulans , B. macerans , alkalophilic Bacillus sp., Pseudomonas , among others) was published in 1992 [5]. The authors analyzed the action pattern of these enzymes and found that they could open CDs to lead to the corresponding OM. For example, the action of Pseudomonas α-amylase on α-CD results in a very rapid accumulation of maltohexaose, but then the latter is degraded to smaller molecules, mainly maltotriose, maltose and glucose. A similar behavior was reported by H. Bender for a CD-degrading enzyme isolated from Flavobacterium sp. The enzyme hydrolyzed OM and CDs to glucose, maltose, and maltotriose [6].
Uchida et al. [7] reported the use of cyclomaltodextrinase from Bacillus sphaericus E-244 for the preparation of maltoheptaose and maltooctaose from β-CD and γ-CD respectively. The enzyme has not only a CD-hydrolyzing activity (decycling of CDs) but also a coupling activity (transfer of a D-glucose unit). They described the enzymatic synthesis of maltoocta- and nonaose from CDs and D-glucose by the coupling reaction, catalyzed by cyclomaltodextrinase ( Scheme 2 ). However, the preparation of maltohexa-, hepta- and/or octaose by this approach required further purification, because small molecules such as glucose, maltose, and maltotriose were also formed.
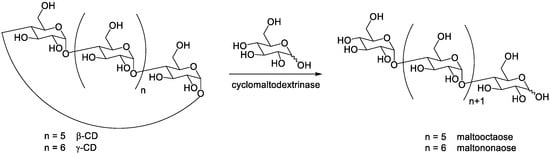
These limitations were circumvented by Fraschini et al. [8] by using modified CDs in one primary position (oxidized to carboxylic acid) and two different enzymes: CD glucanotransferase (CGTase) and amyloglucosidase. They obtained mainly tri- and tetrasaccharide derivatives, thus avoiding further degradation.
PFTA, a thermostable amylase from Pyrococcus furiosus, was cloned and expressed in Escherichia coli. Unlike CD-hydrolyzing enzymes, which were known to produce mainly maltose starting from CDs, PFTA liberated various small OM (from mono- to heptasaccharide) [9]. The difference in the rate of CDs opening (fast) and the hydrolytic activity on the resulting open-chain OM (slow) was exploited to obtain mixtures rich in maltohexaose, maltoheptaose, and maltooctaose from α-, β-, and γ-CD respectively ( Scheme 3 ) [10].
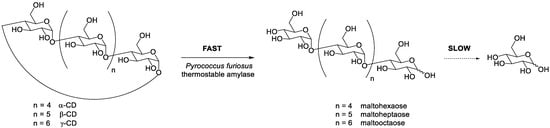
3. Acetolytic Cleavage of Cyclodextrins
The viability of the acetolytic cleavage process in the production of OM from CD derivatives has evolved since the middle of the last century [11][12][13][14]. The early efforts to selectively prepare these oligomers were unsuccessful and led to a mixture of mono and oligomers, in which the amount of the desired hexa-, hepta-, or octa- maltooligosaccharides did not exceed 2–4% [11][12][13][14]. In 1990, Lipták et al. [15] patented their research results on the acetolysis of peracetylated CDs using a mixture of acetic anhydride and sulfuric acid. To increase the efficiency of previous procedures, they optimized the reaction conditions by varying the amount of sulfuric acid between 1–10% in acetic anhydride, the temperature (0–100 °C), and the reaction time [15]. The Table 1 summarizes the results for the acetolysis of peracetylated α-CD.
| Entry | Temperature (°C) | Time (h) | Sulfuric Acid Conc. (vol%) | Yield % |
|---|---|---|---|---|
| 1 | 0 | 72 | 6 | 35 |
| 2 | 30 | 30 | 6 | 35 |
| 3 | 40 | 11 | 6 | 38 |
| 4 | 50 | 10 | 2 | 35 |
| 5 | 50 | 4 | 6 | 49 |
| 6 | 50 | 1.5 | 10 | 35 |
| 7 | 60 | 2 | 6 | 43 |
| 8 | 78 | 0.5 | 6 | 40 |
| 9 | 100 | 0.1 | 6 | 40 |
During this process, the fission of a single glycosidic bond was observed and two acetyl groups were introduced, one at the reducing end and another at the non-reducing end of the linear OM. The maltoheptamer and maltooctamer were also prepared from peracetyl-β- and γ-CDs in 41% and 52 %, respectively.
In another study, Sakairi et al. [16] extended this method to perbenzoylated CDs to prepare partially benzoylated OM derivatives having acetyl groups at reducing and non-reducing ends. These derivatives are useful for the preparation of various derivatives substituted at both ends. Starting from perbenzoylated CDs and using a similar procedure [16] gave 1I,4VI-di-O-acetyl derivative of octadeca-O-benzoyl-α-maltohexaose, 1I,4VI-di-O-acetylated heneicosa-O-benzoyl-α-maltoheptaose, and tetracosa-O-benzoyl-α-maltooctaose in 51%, 37%, and 48% yield respectively ( Scheme 4 ). Other attempts were performed with chloroacetic anhydride or trifluoroacetic anhydride in presence of a catalytic amount of sulfuric acid. However, these attempts failed, leading to complex mixtures. The authors assumed that this was due to the random cleavage of the glycosidic bond and the intra- or inter-molecular migration of acetyl groups.
More recently, Djedaïni-Pilard et al. [17] significantly improved this procedure. In their conditions, they described the formation of di-O-acetylated maltohexaose, -heptaose, and -octaose derivatives from per-O-benzoylated α- , β-, and γ-CD in 70–82% yields. Thus, the authors extended this method to the ring-opening of CDs, halogenated in primary positions, to lead to novel C-6 modified OMs.
4. Ring-Opening Cyclodextrins Using Other Acidic Conditions
With a pKa of -8, perchloric acid HClO4 is a stronger acid than sulfuric acid. In 2001, with the aim of preparing specific neocyclodextrins (neoCDs), Vasella et al. [18] performed the ring-opening of peracetylated α-CD, carrying out acetolysis by a method derived from that developed by Lipták et al. [15]. This procedure consisted in reacting peracetylated α-CD with Ac2O, in the presence of HClO4 (70% by weight aqueous solution) at 0 °C for 20 h. After crystallization in ethanol, they obtained the desired peracetylated maltohexaose in 95% yield (α/β > 9/1) ( Scheme 8 ). This reaction was confirmed in 2010 by Lehn et al. [19], which described similar yields.
Since the use of perchloric acid led to a better yield than in the case of sulfuric acid, the following year the authors described the application of this new procedure to peracetylated γ-CD. [20] Under these conditions, acetylated maltooctaose was obtained in 80% yield, again after crystallization in ethanol.
Very recently, our group [21] demonstrated the interest of triflic anhydride in DCM, instead of acetic anhydride with sulfuric acid, to perform the ring-opening of α-, β-, and γ-CD. These conditions allow the selective preparation of 1-azido derivatives, propargyl, and allyl glycosides by the opening of the corresponding perbenzoylated CD followed by their in situ glycosylation with rather good yields and selectivity ( Scheme 12 ).
The Table 2 resumes the various conditions of acetolysis or hydrolysis for ring-opening of CD in literature. To highlight the most important parameter of the reaction, we sorted the conditions by type of acid, then by functional groups present on CDs, then by type of CD, and finally by acid concentration. We can thus see that, together with the acid concentration, the temperature and the stirring time are also important factors. We can conclude that perchloric acid at 0 °C led to the highest yields (95% of maltohexaose) from peracetylated α-CD. To avoid esterification or etherification of the cyclodextrin prior to the ring-opening, FeCl3 conditions are very efficient because they led to 20–23% yield of 99% pure peracetylated maltooligosaccharides after three steps of recrystallization, whatever the CD.
| Acid. | [Acid] | Cd | [cd] | Functional Groups | T °C | Time | α/β | Yield | Starting Material Recovered | Ref | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| C-2 et C-3 | C-6 | ||||||||||
| FeCl3 | 0.074 M | α | 0.3 M | none | RT then 70 °C | 2.5 + 3.5h | / | 20% | / | [22] | |
| 0.074 M | β | 0.3 M | none | RT then 70 °C | 2.5 + 3.5h | 9:1 | 22% | / | [22] | ||
| 0.074 M | γ | 0.3 M | none | RT then 70 °C | 2.5 + 3.5h | / | 23.5% | / | [22] | ||
| H2SO4 | 0.373 M | α | / | Ac | 50–60 °C | 20 h | 5.3:1 | 47% | 46% | [23] | |
| 1.2 M | α | / | Ac | 50 °C | 4 h | / | 48% | / | [15] | ||
| 0.373 M | β | / | Ac | 50–60 °C | 20 h | / | 41% | 49% | [23] | ||
| 0.373 M | γ | / | Ac | 50–60 °C | 20 h | / | 52% | 37% | [23] | ||
| 0.560 M | β | 0.021 M | Ac | Br | 57 °C | 28 h | / | 16% | 78% | [17] | |
| 0.373 M | β | 0.043 M | Bz | 1 I and 6 Bz | 55 °C | 24 h | / | 70% * | 9% | [24] | |
| 0.373 M | β | 0.044 M | Bz | 1 N3 and 6 Bz | 55 °C | 24 h | / | 73% | 10% | [24] | |
| 0.666 M | β | 0.099 M | Bz | 57 °C | 30 h | / | 32% | 58% | [17] | ||
| 0.373 M | α | 0.035 M | Bz | 60 °C | 30 h | / | 82% | 15% | [17] | ||
| 1.373 M | α | 0.036 M | Bz | 50 °C | 32 h | / | 51% | 36% | [16] | ||
| 0.373 M | β | 0.080 M | Bz | 55 °C | 42 h | / | 76% | 12% | [17] | ||
| 1.373 M | β | 0.086 M | Bz | 50 °C | 29 h | / | 37% | 54% | [16] | ||
| 0.373 M | γ | 0.015 M | Bz | 50 °C | 35 h | / | 70% | 17% | [17] | ||
| 1.373 M | γ | 0.015 M | Bz | 50 °C | 27 h | / | 48% | 39% | [16] | ||
| 0.373 M | β | 0.102 M | Bz | N3 | 55 °C | 30 h | / | 30% | 66% | [17] | |
| HClO4 | 0.086 M | α | 0.019 M | Ac | 0 °C | 22 h | / | 60% | 30% | [17] | |
| 0.087 M | α | 0.019 M | Ac | 0 °C | 45 h | >9:1 | 95% | / | [18] | ||
| 0.087 M | α | 0.019 M | Ac | 0 °C | 45 h | >9:1 | 95% | / | [18] | ||
| 0.084 M | β | 0.018 M | Ac | 0 °C | 20 h | / | 35% | 55% | [17] | ||
| 0.086 M | β | / | Bz | Br | 0 °C then 36 °C | 2 × 20 h | / | 30% | / | [17] | |
| 0.036 M | β | 0.0072 M | Bz | N3 | −20 °C | 16 h | / | 85% | / | [25] | |
| ZnBr(+PhSTMS) | 0.266 M | α | 0.066 M | Me | RT | 5 days | 1:1 | 28% | 68% | [26] | |
| 0.2 M | β | 0.2 M | Me | RT | 4 days | 1:1 | 40% | / | [26] | ||
| 0.2 M | γ | 0.05 M | Me | RT | 4 days | 2:8 | 41% | / | [26] | ||
| TiCl4 | 0.4 M | α | 0.1 M | Me, Et or All | 10 °C | 45h | 96:4 | 41% * | / | [27] | |
| 0.4 M | β | 0.1 M | Me, Et or All | RT | 24h | 99:1 | 66% * | / | [27] | ||
| 0.4 M | γ | 0.1 M | Me, Et or All | 10 °C | 45h | 99:1 | 92%* | / | [27] | ||
References
- Takahashi, Y.; Ogawa, T. Total Synthesis of Cyclomaltohexaose. Carbohydr. Res. 1987, 164, 277–296.
- Takahashi, Y.; Ogawa, T. Total Synthesis of Cyclomaltooctaose and an Isomer of Cyclomaltohexaose, Cyclo{→6)-[α-d-Glcp-(1→4)]5-α-d-Glcp-(1-}. Carbohydr. Res. 1987, 169, 127–149.
- Crini, G. Review: A History of Cyclodextrins. Chem. Rev. 2014, 114, 10940–10975.
- Larsen, D.; Beeren, S.R. Building up Cyclodextrins from Scratch—Templated Enzymatic Synthesis of Cyclodextrins Directly from Maltose. Chem. Commun. 2021, 57, 2503–2506.
- Saha, B.C.; Zeikus, J.G. Cyclodextrin Degrading Enzymes. Starch-Stärke 1992, 44, 312–315.
- Bender, H. Purification and Characterization of a Cyclodextrin-Degrading Enzyme from Flavobacterium sp. Appl. Microbiol. Biotechnol. 1993, 39, 714–719.
- Uchida, R.; Nasu, A.; Tobe, K.; Oguma, T.; Yamaji, N. A Convenient Preparation of Maltooctaose and Maltononaose by the Coupling Reaction of Cyclomaltodextrinase. Carbohydr. Res. 1996, 287, 271–274.
- Fraschini, C.; Greffe, L.; Driguez, H.; Vignon, M.R. Chemoenzymatic Synthesis of 6ω-Modified Maltooligosaccharides from Cyclodextrin Derivatives. Carbohydr. Res. 2005, 340, 1893–1899.
- Yang, S.-J.; Lee, H.-S.; Park, C.-S.; Kim, Y.-R.; Moon, T.-W.; Park, K.-H. Enzymatic Analysis of an Amylolytic Enzyme from the Hyperthermophilic Archaeon Pyrococcus Furiosus Reveals Its Novel Catalytic Properties as Both an Alpha-Amylase and a Cyclodextrin-Hydrolyzing Enzyme. Appl. Environ. Microbiol. 2004, 70, 5988–5995.
- Yang, S.-J.; Lee, H.-S.; Kim, J.-W.; Lee, M.-H.; Auh, J.-H.; Lee, B.-H.; Park, K.-H. Enzymatic Preparation of Maltohexaose, Maltoheptaose, and Maltooctaose by the Preferential Cyclomaltooligosaccharide (Cyclodextrin) Ring-Opening Reaction of Pyrococcus Furiosus Thermostable Amylase. Carbohydr. Res. 2006, 341, 420–424.
- French, D.; Levine, M.L.; Pazur, J.H. Studies on the Schardinger Dextrins. II. Preparation and Properties of Amyloheptaose. J. Am. Chem. Soc. 1949, 71, 356–358.
- French, D.; Knapp, D.W.; Pazur, J.H. Studies on the Schardinger Dextrins. VI. The Molecular Size and Structure of the γ-Dextrin1. J. Am. Chem. Soc. 1950, 72, 5150–5152.
- Freudenberg, K. Hydrolysis and Optical Rotation of Cellulose, Starch, and Cycloglucans. J. Polym. Sci. 1957, 23, 791–799.
- Freudenberg, K.; Blomqvist, G.; Ewald, L.; Soff, K. Hydrolyse Und Acetolyse Der Stärke Und Der Schardinger-Dextrine. Chem. Ber. 1936, 69, 1258–1266.
- Liptak, A.; Nanasi, P.; Szejtli, J.; Kari, Z.M. Process for Producing Linear Maltooligosaccharides Comprising 6, 7 or 8 Glucose Units and Their Peracetylated Derivatives. Hungarian Patent 204857 B, 28 May 1990.
- Sakairi, N.; Matsui, K.; Kuzuhara, H. Acetolytic Fission of a Single Glycosidic Bond of Fully Benzoylated α-, β-, and γ-Cyclodextrins. A Novel Approach to the Preparation of Maltooligosaccharide Derivatives Regioselectively Modified at Their Nonreducing Ends. Carbohydr. Res. 1995, 266, 263–268.
- Lesur, D.; Gassama, A.; Moreau, V.; Pilard, S.; Djedaïni-Pilard, F. Synthesis of Regioselectively and Uniformly Modified Maltoheptaose Derivatives from Cyclomaltoheptaose Precursors. Carbohydr. Res. 2005, 340, 1225–1231.
- Hoffmann, B.; Zanini, D.; Ripoche, I.; Bürli, R.; Vasella, A. Oligosaccharide Analogues of Polysaccharides, Part 22, Synthesis of Cyclodextrin Analogues Containing a Buta-1,3-Diyne-1,4-Diyl or a Butane-1,4-Diyl Unit. Helv. Chim. Acta 2001, 84, 1862–1888.
- Ruff, Y.; Buhler, E.; Candau, S.-J.; Kesselman, E.; Talmon, Y.; Lehn, J.-M. Glycodynamers: Dynamic Polymers Bearing Oligosaccharides Residues—Generation, Structure, Physicochemical, Component Exchange, and Lectin Binding Properties. J. Am. Chem. Soc. 2010, 132, 2573–2584.
- Hoffmann, B.; Bernet, B.; Vasella, A. Oligosaccharide Analogues of Polysaccharides. Helv. Chim. Acta 2002, 85, 265–287.
- Pélingre, M.; Smadhi, M.; Bil, A.; Bonnet, V.; Kovensky, J. One-Pot Synthesis of Asymmetrically Difunctionalized Oligomaltosides by Cyclodextrin Ring Opening. ChemistryOpen 2021, 10, 493–496.
- Farkas, E.; Jánossy, L.; Harangi, J.; Kandra, L.; Lipták, A. Synthesis of Chromogenic Substrates of α-Amylases on a Cyclodextrin Basis. Carbohydr. Res. 1997, 303, 407–415.
- Sakairi, N.; Wang, L.-X.; Kuzuhara, H. Insertion of a D-Glucosamine Residue into the α-Cyclodextrin Skeleton; A Model Synthesis of ‘Chimera Cyclodextrins’. J. Chem. Soc. Chem. Commun. 1991, 5, 289–290.
- Lesur, D.; Gassama, A.; Brique, A.; Thiebault, N.; Djedaïni-Pilard, F.; Pilard, S.; Moreau, V. Synthesis and Characterization of Regioselectively Monoderivatized Maltooligosaccharides through a Combination of Tandem Mass Spectrometry and Enzymatic Hydrolysis Studies. Arkivoc 2013, 2, 276–289.
- Almant, M.; Moreau, V.; Kovensky, J.; Bouckaert, J.; Gouin, S.G. Clustering of Escherichia Coli Type-1 Fimbrial Adhesins by Using Multimeric Heptyl α-D-Mannoside Probes with a Carbohydrate Core. Chem. Eur. J. 2011, 17, 10029–10038.
- Sakairi, N.; Kuzuhara, H. Facile Preparation of Phenyl 1-Thioglycosides of Partially Methylated Maltooligosaccharides by Restricted Thiolysis of Fully Methylated Cyclodextrins. Carbohydr. Res. 1996, 280, 139–143.
- Bösch, A.; Mischnick, P. Bifunctional Building Blocks for Glyco-Architectures by TiCl4-Promoted Ring Opening of Cyclodextrin Derivatives. Biomacromolecules 2007, 8, 2311–2320.

