 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Martina Capelletti | + 2210 word(s) | 2210 | 2020-07-14 05:14:45 | | | |
| 2 | Martina Capelletti | Meta information modification | 2210 | 2020-07-17 17:15:27 | | | | |
| 3 | Martina Capelletti | -1 word(s) | 2209 | 2020-07-17 17:19:38 | | | | |
| 4 | Martina Capelletti | -1 word(s) | 2209 | 2020-07-17 17:22:17 | | | | |
| 5 | Martina Capelletti | Meta information modification | 2209 | 2020-07-17 17:39:55 | | | | |
| 6 | Catherine Yang | -1 word(s) | 2208 | 2020-07-21 06:32:37 | | |
Video Upload Options
Ferroptosis is an iron-dependent form of cell death characterized by intracellular lipid peroxide accumulation and redox imbalance. Ferroptosis shows specific biological and morphological features when compared to the other cell death patterns. The loss of lipid peroxide repair activity by glutathione peroxidase 4 (GPX4), the presence of redox-active iron and the oxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids are considered as distinct fingerprints of ferroptosis. Several pathways, including amino acid and iron metabolism, ferritinophagy, cell adhesion, p53, Keap1/Nrf2 and phospholipid biosynthesis, can modify susceptibility to ferroptosis. Through the decades, various diseases, including acute kidney injury; cancer; ischemia-reperfusion injury; and cardiovascular, neurodegenerative and hepatic disorders, have been associated with ferroptosis. Here, we provide a short overview of the main biological and biochemical mechanisms of ferroptosis. The contribution of ferroptosis to the spectrum of liver diseases, acute or chronic is also reported. Finally, we discuss the use of ferroptosis as a therapeutic approach against hepatocellular carcinoma, the most common form of primary liver cancer.
1. Definition
Ferroptosis is an iron-dependent form of cell death characterized by intracellular lipid peroxide accumulation and redox imbalance. Ferroptosis shows specific biological and morphological features when compared to the other cell death patterns.
2. Discovery of Ferroptosis
In 2003, a study was conducted to identify new molecules with lethal effects on Ras-mutated cells. Among the 23,550, different chemical compounds screened, NSC146109, renamed erastin, had a selectively lethal effect on Ras-expressing cancer cells [1]. In 2008, two other molecules, Ras-selective poisonous small molecules (RSL3-5), were identified, which kill selectively human foreskin fibroblasts (BJeLR) in a non-apoptotic manner [2]. Inhibitors specific for RCDs cannot undo RSL-induced cell death. In contrast, antioxidants (vitamin E) and iron chelators can block and reverse RSL-induced cell death [3]. Therefore, the term “ferroptosis” refers to an iron-dependent, non-apoptotic cell death characterized by lipid peroxidation [4].
3. Mechanisms of Ferroptosis
Ferroptosis is a sum of many biological pathways, acting simultaneously. In Figure 1, ferroptosis regulatory pathways are reported. Three main biological axes are roughly:
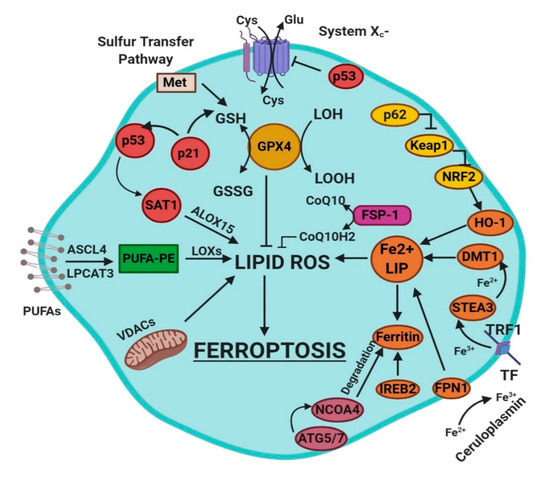
Figure 1. This figure summarizes the regulatory core of ferroptosis, approximately divided into three axes. The first axis is represented in the middle. It includes the GSH/GPX4, sulfur transfer and p53 pathways. The second axis (right part of theF) consists of the iron metabolism pathway, including IREB2 related to ferritin metabolism, the regulation of ATG5-ATG7-NCOA4 pathway and the p62-Keap1-Nrf2 regulatory pathway. These elements can influence the concentration of intracellular iron, mandatory for the development of ferroptosis. On the left, the third axis implies that lipid metabolism p53-spermidine/spermine N1-acetyltransferase 1 (SAT1)-ALOX15, ACSL4, LPCAT3, etc. impact on fatty acids regulation and ferroptosis [4]. Finally, mitochondria are also involved, since VDACs (voltage-dependent anion channels) are inhibited by erastin. In parallel, the independent pathway ferroptosis suppressor protein 1-coenzyme Q10 (FSP-1-CoQ10) acts with GSH/GPX4 to contrast lipid peroxidation [4].
-
glutathione/glutathione peroxidase 4 (GSH/GPX4) pathway, inhibition of system Xc−, sulfur transfer pathway, and p53 regulatory axis;
-
iron metabolism with the regulation of autophagy protein 5 and 7 (ATG5-ATG7) and nuclear receptor coactivator 4 (NCOA4) pathway and iron-responsive element-binding protein 2 (IREB2) related to ferritin metabolism, and the p62-Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor (Nrf2) regulatory pathways [4];
-
lipid metabolism pathways as p53, arachidonate lipoxygenase 15 (ALOX15), acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3) [4].
4. Ferroptosis in Liver Diseases
Hepatocytes play a crucial role in humans by helping maintain stable glucose and lipoprotein concentrations in the plasma. Usually, hepatocytes are quiescent, but a radical change in liver physiology can occur when liver tissue is exposed to viruses, toxic agents or metabolites in excess. Moreover, hepatocytes are the primary site of the storage of iron in the body. Clinicians set 13–15 mg of iron/g of liver tissue as a critical threshold, which is associated with an increased risk of cirrhosis. [5]. The type of liver damage depends on the nature and the severity of the lesion. Different kinds of RCDs may coexist in the progression of metabolic liver diseases to inflammation, fibrosis and, ultimately, cirrhosis [6]. Cirrhosis, a slow process spread over decades, is the most advanced stage of liver fibrosis and is associated with a higher risk of malignant liver transformation into hepatocellular carcinoma (HCC). Accumulating evidence suggests that lytic cell death modalities (e.g., necroptosis, pyroptosis and ferroptosis) elicit strong inflammatory responses due to cell membrane permeabilization and release of cellular components, contributing to the recruitment of immune cells and activation of hepatic stellate cells [6].
The association between liver damage and both inherited and acquired iron overload is indisputable. Ferroptosis could determine iron overload because the induction of ferritinophagy induces the active mobilization of cellular iron. In any case, uncontrolled free iron exerts a toxic effect on the liver, stimulating the advancement of hepatic diseases and leading to severe collateral effects [7].
4.1. Ferroptosis and Drug-Induced Liver Injury
Drug-induced liver injury (DILI) is the predominant cause of acute liver diseases (ALD) in Europe and the USA, with acetaminophen as the paradigmatic example [8]. The exposure of hepatic tissue to acetaminophen leads to the hepatocyte cell death [9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][5][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][8][52]. Its transformation by cytochrome p450 provokes liver-toxicity through its reactive metabolite NAPQI (N-acetyl-p-benzoquinone imine) [52]. NAPQI binds to GSH and leads to severe depletion of GSH in hepatocytes [52]. Yamada’s team has recently found that ferroptosis driven by ω-6 PUFAs is associated with acetaminophen-induced ALD [53]. Besides, ferrostatin-1, DFO and vitamin E could exert a protective effect on hepatocytes by suppressing lipid peroxidation and GSH depletion [53]. Moreover, it was recently shown using CRISPR-Cas9 that cytochrome P450 oxidoreductase, which is directly implied in the detoxification of xenobiotics by hemoprotein, was necessary for ferroptotic cell death by upregulating the PUFAs peroxidation [54].
4.2. Ferroptosis and Ischemia–Reperfusion Injury (IRI)
IRI is the consequence of a temporary reduction of the blood supply followed by revascularization [55]. The restoration of blood supply after ischemia aggravates the pre-existing injury in the liver caused by ischemia. Liver IRI can be induced by shock (e.g., sepsis and hemorrhage) or after liver surgery. In 2014, Friedmann Angeli et al., hypothesized a protective effect of liproxstatin-1 against hepatic damage in a mouse model of hepatic injury induced by IRI [25]. These data evidenced that ferroptosis was a mechanism implied in liver IRI, underlying the potential interest to target ferroptosis in the treatment of IRI (Figure 2).
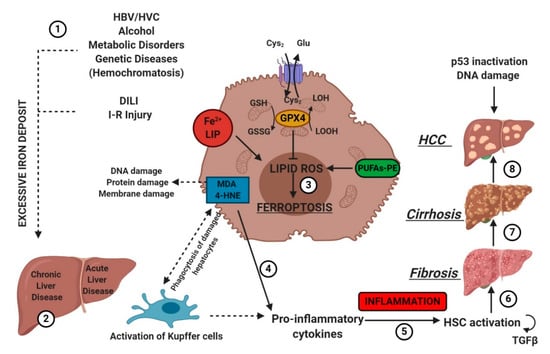
Figure 2. Etiologic factors and the central role of oxidative stress in the development of hepatic diseases. Several factors (e.g., HBV/HCV infection, inadequate alcohol or drug consumption, metabolic or genetic diseases, and I-R injury (1)) may trigger CLD or ALD (2) in the liver, promoting iron accumulation. This supports pro-oxidative state triggering protein and DNA damage and lipid peroxidation (3). The instauration of hepatocyte oxidative stress conditions results in inflammation (4–5), which favors liver fibrosis (6) and cirrhosis (7), and it may lead to hepatocellular carcinoma (8). Numbers (1–8) were added to indicate the path to follow in reading the image. HSC, hematopoietic stem cells; TGFβ, transforming growth factor-beta.
4.3. Chronic Liver Diseases (CLD)
CLD are the consequence of constant exposure of the liver to agents as alcohol, chronic infection such as HBV/HVC or altered intermediary metabolism [9][10][11][12][13][14][15][16][17][18][19][20][21][22][23][24][25][26][27][5][28][29][30][31][32][33][34][35][36][37][38][39][40][41][42][43][44][45][46][47][48][49][50][51][8][52]. These conditions induce chronic liver scarring and fibrosis, combined with an increased cell turnover rate.
CLD and iron metabolism are tightly interconnected. Chronic HVC infection favors the accumulation of iron, downregulating hepatic hepcidin and upregulating duodenal ferroportin-1. Progressive iron overload correlates with the worsening of liver damage.
An iron-centered hypothesis has also been proposed in the pathogenesis of the non-alcoholic liver disease (NAFLD) [50]. NAFLD spans from simple hepatic steatosis to non-alcoholic steatohepatitis (NASH), and it is associated with obesity and metabolic syndrome [50]. Steatosis can progress to an inflammatory state known as non-alcoholic steatohepatitis (NASH) and degenerate to fibrosis. This progression correlates with an increase in lipid peroxidation, stemming from the simultaneous presence of fatty acids and a high concentration of iron in hepatocytes [56]. Lipotoxicity, oxidative stress, organelle dysfunction and inflammatory response exacerbate ballooning and increase hepatocyte cell death [57]. The role of ferroptosis in NAFLD and NASH has not been clarified yet, but MDA and 4-HNE have been used as oxidative stress markers in NASH patients [58]. Besides, the hepatic PC/PE ratio is low in NASH patients. Vitamin E suppresses lipid peroxidation and reduces serum transaminases in NASH patients [59]. Conversely, iron overload aggravates NASH [60][61]. Ferroptosis is the first hit event that triggers steatohepatitis and precedes other RCDs. [62]. Ferroptosis inhibitors, as Trolox and deferiprone, suppress cell death and inflammation at the onset of NASH in the choline-deficient, ethionine-supplemented (CDE) diet model. In alcohol liver disease, after ethanol administration, adipose-specific lipin-1 overexpression accelerates iron accumulation, causes lipid peroxidation, reduces GSH and GAPDH and promotes ferroptotic liver damage in mice [63]. Lipin-1 is an Mg2+-dependent phosphatidic acid phosphohydrolase, implicated in the formation of diacylglycerol during the synthesis of phospholipids and triglycerides.
Regarding hereditary hemochromatosis, inherited mutations in the HFE gene lead to massive accumulation of iron in the liver and chronic tissue damage that may progress to hepatocellular carcinoma. In 2017, Wang’s team induced ferroptosis in the liver by administering ferric citrate to –SCL7A11 KO mice lacking xCT of system Xc− [64]. Indeed, –SCL7A11 KO facilitates iron overload-induced ferroptosis due to impaired cystine uptake and increased ROS production. Ferrostatin-1 reversed the iron-overload damage [64].
In addition, the rupture of the gene encoding Nrf2 stimulates the onset of liver fibrosis in Hfe−/− by an increased susceptibility to oxidative stress [65].
4.4. Hepatocellular Carcinoma (HCC)
HCC is the most frequent liver cancer, and it is the second leading cause of cancer-related death worldwide in men [66]. HCC is usually diagnosed late. Sorafenib, a multikinase inhibitor, was the first drug used for the treatment of advanced HCC. Louandre et al., evidenced that sorafenib induces ferroptosis [47]. Since then, sorafenib has been classified as a ferroptosis inducer [67]. Sorafenib exerts its anticancer activity by inducing apoptosis and inhibiting proliferation as well as angiogenesis. It is a weak apoptosis inducer compared to other chemicals, but it produces ferroptosis. Sorafenib-induced ferroptosis can be reversed using either ferrostatin-1 or shRNA. Mechanistically, sorafenib acts as erastin inhibiting SCL7A11, blocking the import of oxidized cysteine into the cell. Many studies have been done to characterize the relationship between sorafenib and cellular pathways. The retinoblastoma protein (Rb) inhibits sorafenib-induced ferroptosis in HCC cells [68]. Therefore, the Rb status of individual HCC patients is an important prognostic marker during treatment with sorafenib.
The p62-Keap1-Nrf2 pathway also plays a central role in the prevention of HCC by acting against sorafenib-induced ferroptosis by upregulating genes (notably –HO-1 and FTH1) involved in iron and ROS metabolism [33]. These genes act as antioxidants, enhancing the resistance to ferroptosis. Another protein involved in sorafenib resistance is the metallothionein-1G (MT-1G), a critical negative regulator of ferroptosis. Its expression is induced by sorafenib and other kinase inhibitors (e.g., erlotinib and gefitinib) [67]. The upregulation of MT-1G contributes to sorafenib resistance. Besides, CISD1 negatively regulates ferroptosis, and it is upregulated in an iron-dependent manner in human HCC cells. Finally, a recent study showed that a variant present in Africans and the Afro-American population in the tumor suppressor p53 (p.Pro47Ser) downregulates ferroptosis in HCC [67].
Interestingly, Ou et al., reported that low-density lipoprotein-docosahexaenoic acid (LDL-DHA) nanoparticles selectively kill human and rat hepatoma cell lines in vitro and reduce the growth of liver cancer in rat [69]. LDL-DHA exacerbates lipid peroxidation and determines the depletion of GSH and inactivation of GPX4 before cell death. DHA is responsible for the direct degradation of GPX4, while the entire nanoparticle decreases the concentration of intracellular GSH by reducing redox couples GSH/GSSG and NADPH/NADP+ and removing GSH-aldehyde adducts [66]. In 2017, Bai et al., identified haloperidol as a potential pharmacological modulator. Haloperidol, which is antipsychotic medication and a sigma receptor 1 antagonist, was able to promote erastin- and sorafenib-induced cell death at a relatively low dose [70], indicating that haloperidol may benefit HCC patients treated with sorafenib by reducing the dosage or potentiating its effectiveness [66]. After the administration of haloperidol, the distinct traits (iron accumulation, lipid peroxidation and GSH depletion) of ferroptosis were reported; at the same time, haloperidol influenced ferroptosis-related targets (Nrf2, HO-1 and GPX4) [70]. These results provide a novel pharmacological strategy for HCC therapy.
5. Conclusion and Perspectives
Nowadays, the idea of ferroptosis-inducing therapy is becoming consistent in the field of cancer treatments. Sorafenib is now the gold standard as a ferroptosis inducer. Moreover, new pharmacological formulations are becoming available for erastin and RSL3. Efforts have been undertaken to render erastin more suitable for in vivo applications, such as erastin-loaded exosomes, to target triple-negative breast cancer [71]. New studies focused on the molecular basis of ferroptosis could help in deciphering the existing connections between the single components of the metabolic pathway. Above all, this could also lead to the identification of new molecular markers for ferroptosis, which are still missing. These markers could also lead to a faster diagnosis in the field of oncology. In this context, ferroptosis may represent the cornerstone for the development of alternative curative approaches. In summary, new projects dedicated to fundamental research should be encouraged to obtain the full picture of intracellular interactions after ferroptosis induction. Another field of interest is to define the connections between ferroptosis and other cell deaths to identify their co-operation and modulation in cells and tissues. New frontiers are opening, which will put ferroptosis under the spotlight of future translational medicine.
References
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296.
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379.
- Doll, S.; Conrad, M. Iron and ferroptosis: A still ill-defined liaison: Iron and Ferroptosis’Ferroptosis’. IUBMB Life 2017, 69, 423–434.
- Li, J.; Cao, F.; Yin, H.-L.; Huang, Z.-J.; Lin, Z.-T.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, present and future. Cell Death Dis. 2020, 11, 1–13.
- Piperno, A.; Pelucchi, S.; Mariani, R. Inherited iron overload disorders. Transl. Gastroenterol. Hepatol. 2020, 5, 25.
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M.P. Lytic cell death in metabolic liver disease. J. Hepatol. 2020, 0168827820302105.
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651.
- Kullak-Ublick, G.A.; Andrade, R.J.; Merz, M.; End, P.; Benesic, A.; Gerbes, A.L.; Aithal, G.P. Drug-induced liver injury: Recent advances in diagnosis and risk assessment. Gut 2017, 66, 1154–1164.
- Tang, D. (Ed.) Ferroptosis in Health and Disease; Springer: Cham, Switzerland, 2019.
- Ursini, F.; Maiorino, M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free. Radic. Biol. Med. 2020, 152, 175–185.
- Conrad, M.; Sato, H. The oxidative stress-inducible cystine/glutamate antiporter, system x c − : Cystine supplier and beyond. Amino Acids 2011, 42, 231–246.
- Newstead, S. Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transporters. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 488–499.
- Bannai, S.; Kitamura, E. Transport Interactionof t-Cystine and L-Glutamatein Human Diploid Fibroblasts in Culture. Biol. Chem. 1980, 255, 2372–2376.
- Lewerenz, J.; Hewett, S.; Huang, Y.; Lambros, M.; Gout, P.W.; Kalivas, P.W.; Massie, A.; Smolders, I.; Methner, A.; Pergande, M.; et al. The Cystine/Glutamate Antiporter System xc− in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxidants Redox Signal. 2013, 18, 522–555.
- Lee, J.; Kang, E.S.; Kobayashi, S.; Homma, T.; Sato, H.; Seo, H.G.; Fujii, J. The viability of primary hepatocytes is maintained under a low cysteine-glutathione redox state with a marked elevation in ophthalmic acid production. Exp. Cell Res. 2017, 361, 178–191.
- Hayano, M.; Yang, W.S.; Corn, C.K.; Pagano, N.C.; Stockwell, B.R. Loss of cysteinyl-tRNA synthetase (CARS) induces the transsulfuration pathway and inhibits ferroptosis induced by cystine deprivation. Cell Death Differ. 2015, 23, 270–278.
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311.
- Yant, L.; Ran, Q.; Rao, L.; Van Remmen, H.; Shibatani, T.; Belter, J.G.; Motta, L.; Richardson, A.; Prolla, T.A. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free. Radic. Biol. Med. 2003, 34, 496–502.
- Scheerer, P.; Borchert, A.; Kraus, N.; Wessner, H.; Gerth, C.; Höhne, W.; Kuhn, H. Structural Basis for Catalytic Activity and Enzyme Polymerization of Phospholipid Hydroperoxide Glutathione Peroxidase-4 (GPx4)†,‡,§. Biochemistry 2007, 46, 9041–9049.
- Gaschler, M.M.; Andia, A.A.; Liu, H.; Csuka, J.M.; Hurlocker, B.; Vaiana, C.A.; Heindel, D.W.; Zuckerman, D.S.; Bos, P.H.; Reznik, E.; et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515.
- Tarangelo, A.; Dixon, S.J. Lipid Metabolism and ‘Ferroptosis’. In Ferroptosis in Health and Disease; Tang, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 1–26.
- Lei, P.; Bai, T.; Sun, Y. Mechanisms of Ferroptosis and Relations With Regulated Cell Death: A Review. Front. Physiol. 2019, 10, 139.
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98.
- Feng, H.; Stockwell, B.R. Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol. 2018, 16, e2006203.
- Angeli, J.P.F.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191.
- Gaschler, M.M.; Hu, F.; Feng, H.; Linkermann, A.; Min, W.; Stockwell, B.R. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem. Biol. 2018, 13, 1013–1020.
- Wong-Ekkabut, J.; Xu, Z.; Triampo, W.; Tang, I.-M.; Tieleman, D.P.; Monticelli, L. Effect of Lipid Peroxidation on the Properties of Lipid Bilayers: A Molecular Dynamics Study. Biophys. J. 2007, 93, 4225–4236.
- Camaschella, C.; Nai, A.; Silvestri, L. Iron metabolism and iron disorders revisited in the hepcidin era. Haematologica 2020, 105, 260–272.
- Daher, R.; Manceau, H.; Karim, Z. Iron metabolism and the role of the iron-regulating hormone hepcidin in health and disease. La Presse Médicale 2017, 46, e272–e278. Kwon, M.-Y.; Park, E.; Lee, S.-J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403.
- Sun, X.; Ou, Z.; Xie, M.; Kang, R.; Fan, Y.; Niu, X.; Wang, H.; Cao, L.; Tang, D. HSPB1 as a novel regulator of ferroptotic cancer cell death. Oncogene 2015, 34, 5617–5625.
- Hirayama, T.; Miki, A.; Nagasawa, H. Organelle-specific analysis of labile Fe(ii) during ferroptosis by using a cocktail of various colour organelle-targeted fluorescent probes. Metallomics 2019, 11, 111–117.
- Wang, S.-J.; Li, D.; Ou, Y.; Jiang, L.; Chen, Y.; Zhao, Y.; Gu, W. Acetylation Is Crucial for p53-Mediated Ferroptosis and Tumor Suppression. Cell Rep. 2016, 17, 366–373.
- Sun, X.; Ou, Z.; Chen, R.; Niu, X.; Chen, D.; Kang, R.; Tang, D. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016, 63, 173–184.
- Suzuki, T.; Motohashi, H.; Yamamoto, M. Toward clinical application of the Keap1–Nrf2 pathway. Trends Pharmacol. Sci. 2013, 34, 340–346.
- Copple, I.M. The Keap1–Nrf2 Cell Defense Pathway–A Promising Therapeutic Target? Adv. Pharmacol. 2012, 63, 43–79.
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109.
- Liu, N.; Lin, X.; Huang, C. Activation of the reverse transsulfuration pathway through NRF2/CBS confers erastin-induced ferroptosis resistance. Br. J. Cancer 2020, 122, 279–292.
- Eleftheriadis, T.; Pissas, G.; Liakopoulos, V.; Liakopoulos, V.; Stefanidis, I. The H2S–Nrf2–Antioxidant Proteins Axis Protects Renal Tubular Epithelial Cells of the Native Hibernator Syrian Hamster from Reoxygenation-Induced Cell Death. Biology 2019, 8, 74.
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692.
- Han, C.; Liu, Y.; Dai, R.; Ismail, N.; Su, W.; Li, B. Ferroptosis and Its Potential Role in Human Diseases. Front. Pharmacol. 2020, 11, 239.
- Gao, M.; Yi, J.; Zhu, J.; Minikes, A.; Monian, P.; Thompson, C.B.; Jiang, X. Role of Mitochondria in Ferroptosis. Mol. Cell 2019, 73, 354–363.
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem. Biophys. Res. Commun. 2016, 478, 838–844.
- McCullough, K.; Bolisetty, S. Ferritins in Kidney Disease. Semin. Nephrol. 2020, 40, 160–172.
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta BBA Gen. Subj. 2017, 1861, 1893–1900.
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109.
- Liang, C.; Zhang, X.; Yang, M.; Dong, X. Recent Progress in Ferroptosis Inducers for Cancer Therapy. Adv. Mater. 2019, 31, e1904197.
- Louandre, C.; Ezzoukhry, Z.; Godin, C.; Barbare, J.-C.; Mazière, J.-C.; Chauffert, B.; Galmiche, A. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int. J. Cancer 2013, 133, 1732–1742.
- Angeli, J.P.F.; Shah, R.; Pratt, D.A.; Conrad, M. Ferroptosis Inhibition: Mechanisms and Opportunities. Trends Pharmacol. Sci. 2017, 38, 489–498.
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent. Sci. 2017, 3, 232–243.
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M.P. Lytic cell death in metabolic liver disease. J. Hepatol. 2020, 0168827820302105.
- Macías-Rodríguez, R.U.; Inzaugarat, M.E.; Ruiz-Margáin, A.; Nelson, L.; Trautwein, C.; Cubero, F.J. Reclassifying Hepatic Cell Death during Liver Damage: Ferroptosis—A Novel Form of Non-Apoptotic Cell Death? Int. J. Mol. Sci. 2020, 21, 1651.
- Galmiche, A. Ferroptosis in Liver Disease. In Ferroptosis in Health and Disease; Tang, D., Ed.; Springer: Cham, Switzerland, 2019; pp. 239–248.
- Yamada, N.; Karasawa, T.; Kimura, H.; Watanabe, S.; Komada, T.; Kamata, R.; Sampilvanjil, A.; Ito, J.; Nakagawa, K.; Kuwata, H.; et al. Ferroptosis driven by radical oxidation of n-6 polyunsaturated fatty acids mediates acetaminophen-induced acute liver failure. Cell Death Dis. 2020, 11, 144.
- Zou, Y.; Li, H.; Graham, E.T.; Deik, A.A.; Eaton, J.K.; Wang, W.; Sandoval-Gomez, G.; Clish, C.; Doench, J.G.; Schreiber, S.L. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat. Chem. Biol. 2020, 16, 302–309.
- Saidi, R.F.; Kenari, S.K.H. Liver Ischemia/Reperfusion Injury: An Overview. J. Investig. Surg. 2014, 27, 366–379.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Afonso, M.B.; Castro, R.E.; Rodrigues, C.M.P. Processes exacerbating apoptosis in non-alcoholic steatohepatitis. Clin. Sci. 2019, 133, 2245–2264.
- Loguercio, C.; De Girolamo, V.; De Sio, I.; Tuccillo, C.; Ascione, A.; Baldi, F.; Budillon, G.; Cimino, L.; Di Carlo, A.; Di Marino, M.P.; et al. Non-alcoholic fatty liver disease in an area of southern Italy: Main clinical, histological, and pathophysiological aspects. J. Hepatol. 2001, 35, 568–574.
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; LaVine, J.E.; Tonascia, J.; Ünalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. New Engl. J. Med. 2010, 362, 1675–1685.
- Nelson, J.E.; Wilson, L.; Brunt, E.M.; Yeh, M.M.; Kleiner, D.E.; Unalp-Arida, A.; Kowdley, K.V.; Nonalcoholic Steatohepatitis Clinical Research Network. Relationship between the pattern of hepatic iron deposition and histological severity in nonalcoholic fatty liver disease. Hepatology 2011, 53, 448–457.
- Bonkovsky, H.L.; Jawaid, Q.; Tortorelli, K.; LeClair, P.; Cobb, J.; Lambrecht, R.W.; Banner, B.F. Non-alcoholic steatohepatitis and iron: Increased prevalence of mutations of the HFE gene in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 421–429.
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.-Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 1–14.
- Zhou, Z.; Ye, T.J.; Bonavita, G.; Daniels, M.; Kainrad, N.; Jogasuria, A.; You, M. Adipose-Specific Lipin-1 Overexpression Renders Hepatic Ferroptosis and Exacerbates Alcoholic Steatohepatitis in Mice. Hepatol. Commun. 2019, 3, 656–669.
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465.
- Duarte, T.L.; Caldas, C.; Santos, A.G.; Silva-Gomes, S.; Santos-Gonçalves, A.; Martins, M.J.; Porto, G.; Lopes, J.M. Genetic disruption of NRF2 promotes the development of necroinflammation and liver fibrosis in a mouse model of HFE-hereditary hemochromatosis. Redox Biol. 2017, 11, 157–169.
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424.
- Nie, J.; Lin, B.; Zhou, M.; Wu, L.; Zheng, T. Role of ferroptosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2018, 144, 2329–2337.
- Louandre, C.; Marcq, I.; Bouhlal, H.; Lachaier, E.; Godin, C.; Saidak, Z.; Francois, C.; Chatelain, D.; DeBuysscher, V.; Barbare, J.-C.; et al. The retinoblastoma (Rb) protein regulates ferroptosis induced by sorafenib in human hepatocellular carcinoma cells. Cancer Lett. 2015, 356, 971–977.
- Ou, W.; Mulik, R.S.; Anwar, A.; McDonald, J.G.; He, X.; Corbin, I.R. Low-density lipoprotein docosahexaenoic acid nanoparticles induce ferroptotic cell death in hepatocellular carcinoma. Free. Radic. Biol. Med. 2017, 112, 597–607.
- Bai, T.; Wang, S.; Zhao, Y.; Zhu, R.; Wang, W.; Sun, Y. Haloperidol, a sigma receptor 1 antagonist, promotes ferroptosis in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2017, 491, 919–925.
- Bebber, C.M.; Müller, F.; Clemente, L.P.; Weber, J.; Von Karstedt, S. Ferroptosis in Cancer Cell Biology. Cancers 2020, 12, 164

