Ferroptosis is an iron-dependent form of cell death characterized by intracellular lipid peroxide accumulation and redox imbalance. Ferroptosis shows specific biological and morphological features when compared to the other cell death patterns. The loss of lipid peroxide repair activity by glutathione peroxidase 4 (GPX4), the presence of redox-active iron and the oxidation of polyunsaturated fatty acid (PUFA)-containing phospholipids are considered as distinct fingerprints of ferroptosis. Several pathways, including amino acid and iron metabolism, ferritinophagy, cell adhesion, p53, Keap1/Nrf2 and phospholipid biosynthesis, can modify susceptibility to ferroptosis. Through the decades, various diseases, including acute kidney injury; cancer; ischemia-reperfusion injury; and cardiovascular, neurodegenerative and hepatic disorders, have been associated with ferroptosis. Here, we provide a short overview of the main biological and biochemical mechanisms of ferroptosis. The contribution of ferroptosis to the spectrum of liver diseases, acute or chronic is also reported. Finally, we discuss the use of ferroptosis as a therapeutic approach against hepatocellular carcinoma, the most common form of primary liver cancer.
- cell death
- ferroptosis
- iron
- iron metabolism
- liver
- hepatic diseases
1. Definition
Ferroptosis is an iron-dependent form of cell death characterized by intracellular lipid peroxide accumulation and redox imbalance. Ferroptosis shows specific biological and morphological features when compared to the other cell death patterns.
2. Discovery of Ferroptosis
In 2003, a study was conducted to identify new molecules with lethal effects on Ras-mutated cells. Among the 23,550, different chemical compounds screened, NSC146109, renamed erastin, had a selectively lethal effect on Ras-expressing cancer cells [4]. In 2008, two other molecules, Ras-selective poisonous small molecules (RSL3-5), were identified, which kill selectively human foreskin fibroblasts (BJeLR) in a non-apoptotic manner [5]. Inhibitors specific for RCDs cannot undo RSL-induced cell death. In contrast, antioxidants (vitamin E) and iron chelators can block and reverse RSL-induced cell death [1]. Therefore, the term “ferroptosis” refers to an iron-dependent, non-apoptotic cell death characterized by lipid peroxidation [3].
3. Mechanisms of Ferroptosis
Ferroptosis is a sum of many biological pathways, acting simultaneously. In Figure 1, ferroptosis regulatory pathways are reported. Three main biological axes are roughly:
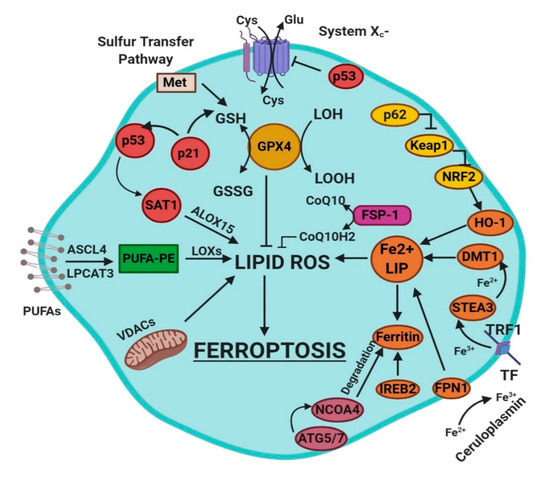
Figure 1. This figure summarizes the regulatory core of ferroptosis, approximately divided into three axes. The first axis is represented in the middle. It includes the GSH/GPX4, sulfur transfer and p53 pathways. The second axis (right part of theF) consists of the iron metabolism pathway, including IREB2 related to ferritin metabolism, the regulation of ATG5-ATG7-NCOA4 pathway and the p62-Keap1-Nrf2 regulatory pathway. These elements can influence the concentration of intracellular iron, mandatory for the development of ferroptosis. On the left, the third axis implies that lipid metabolism p53-spermidine/spermine N1-acetyltransferase 1 (SAT1)-ALOX15, ACSL4, LPCAT3, etc. impact on fatty acids regulation and ferroptosis [3]. Finally, mitochondria are also involved, since VDACs (voltage-dependent anion channels) are inhibited by erastin. In parallel, the independent pathway ferroptosis suppressor protein 1-coenzyme Q10 (FSP-1-CoQ10) acts with GSH/GPX4 to contrast lipid peroxidation [3].
-
glutathione/glutathione peroxidase 4 (GSH/GPX4) pathway, inhibition of system Xc−, sulfur transfer pathway, and p53 regulatory axis;
-
iron metabolism with the regulation of autophagy protein 5 and 7 (ATG5-ATG7) and nuclear receptor coactivator 4 (NCOA4) pathway and iron-responsive element-binding protein 2 (IREB2) related to ferritin metabolism, and the p62-Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor (Nrf2) regulatory pathways [3];
-
lipid metabolism pathways as p53, arachidonate lipoxygenase 15 (ALOX15), acyl-CoA synthetase long-chain family member 4 (ACSL4), lysophosphatidylcholine acyltransferase 3 (LPCAT3) [3].
4. Ferroptosis in Liver Diseases
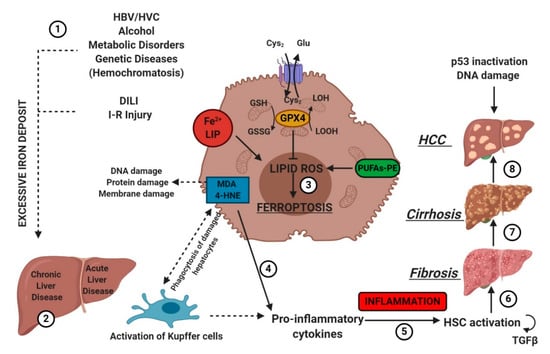
Hepatocytes play a crucial role in humans by helping maintain stable glucose and lipoprotein concentrations in the plasma. Usually, hepatocytes are quiescent, but a radical change in liver physiology can occur when liver tissue is exposed to viruses, toxic agents or metabolites in excess. Moreover, hepatocytes are the primary site of the storage of iron in the body. Clinicians set 13–15 mg of iron/g of liver tissue as a critical threshold, which is associated with an increased risk of cirrhosis. [29]. The type of liver damage depends on the nature and the severity of the lesion. Different kinds of RCDs may coexist in the progression of metabolic liver diseases to inflammation, fibrosis and, ultimately, cirrhosis [53]. Cirrhosis, a slow process spread over decades, is the most advanced stage of liver fibrosis and is associated with a higher risk of malignant liver transformation into hepatocellular carcinoma (HCC). Accumulating evidence suggests that lytic cell death modalities (e.g., necroptosis, pyroptosis and ferroptosis) elicit strong inflammatory responses due to cell membrane permeabilization and release of cellular components, contributing to the recruitment of immune cells and activation of hepatic stellate cells [53].
The association between liver damage and both inherited and acquired iron overload is indisputable. Ferroptosis could determine iron overload because the induction of ferritinophagy induces the active mobilization of cellular iron. In any case, uncontrolled free iron exerts a toxic effect on the liver, stimulating the advancement of hepatic diseases and leading to severe collateral effects [54].
4.1. Ferroptosis and Drug-Induced Liver Injury
Drug-induced liver injury (DILI) is the predominant cause of acute liver diseases (ALD) in Europe and the USA, with acetaminophen as the paradigmatic example [55]. The exposure of hepatic tissue to acetaminophen leads to the hepatocyte cell death [10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56]. Its transformation by cytochrome p450 provokes liver-toxicity through its reactive metabolite NAPQI (N-acetyl-p-benzoquinone imine) [56]. NAPQI binds to GSH and leads to severe depletion of GSH in hepatocytes [56]. Yamada’s team has recently found that ferroptosis driven by ω-6 PUFAs is associated with acetaminophen-induced ALD [57]. Besides, ferrostatin-1, DFO and vitamin E could exert a protective effect on hepatocytes by suppressing lipid peroxidation and GSH depletion [57]. Moreover, it was recently shown using CRISPR-Cas9 that cytochrome P450 oxidoreductase, which is directly implied in the detoxification of xenobiotics by hemoprotein, was necessary for ferroptotic cell death by upregulating the PUFAs peroxidation [58].
4.2. Ferroptosis and Ischemia–Reperfusion Injury (IRI)
IRI is the consequence of a temporary reduction of the blood supply followed by revascularization [59]. The restoration of blood supply after ischemia aggravates the pre-existing injury in the liver caused by ischemia. Liver IRI can be induced by shock (e.g., sepsis and hemorrhage) or after liver surgery. In 2014, Friedmann Angeli et al., hypothesized a protective effect of liproxstatin-1 against hepatic damage in a mouse model of hepatic injury induced by IRI [26]. These data evidenced that ferroptosis was a mechanism implied in liver IRI, underlying the potential interest to target ferroptosis in the treatment of IRI (Figure 5).
4.3. Chronic Liver Diseases (CLD)
4.4. Hepatocellular Carcinoma (HCC)
5. Conclusion and Perspectives
Nowadays, the idea of ferroptosis-inducing therapy is becoming consistent in the field of cancer treatments. Sorafenib is now the gold standard as a ferroptosis inducer. Moreover, new pharmacological formulations are becoming available for erastin and RSL3. Efforts have been undertaken to render erastin more suitable for in vivo applications, such as erastin-loaded exosomes, to target triple-negative breast cancer [75]. New studies focused on the molecular basis of ferroptosis could help in deciphering the existing connections between the single components of the metabolic pathway. Above all, this could also lead to the identification of new molecular markers for ferroptosis, which are still missing. These markers could also lead to a faster diagnosis in the field of oncology. In this context, ferroptosis may represent the cornerstone for the development of alternative curative approaches.
In summary, new projects dedicated to fundamental research should be encouraged to obtain the full picture of intracellular interactions after ferroptosis induction. Another field of interest is to define the connections between ferroptosis and other cell deaths to identify their co-operation and modulation in cells and tissues. New frontiers are opening, which will put ferroptosis under the spotlight of future translational medicine.
This entry is adapted from the peer-reviewed paper 10.3390/ijms21144908