 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Shin-Da Lee | + 1879 word(s) | 1879 | 2021-08-16 10:42:57 | | | |
| 2 | Lindsay Dong | Meta information modification | 1879 | 2021-08-17 04:49:40 | | |
Video Upload Options
Treadmill training attenuated complex I deficits, cytochrome c release, ATP depletion, and complexes II–V abnormalities in Parkinson’s disease. Studies analyzed the neural mitochondrial quality-control, reporting that treadmill exercise improved mitochondrial biogenesis, mitochondrial fusion, and mitophagy in Parkinson’s disease. The hypothesis that treadmill training could attenuate both neural mitochondrial respiratory deficiency and neural mitochondrial quality-control dysregulation in Parkinson’s disease, suggesting that treadmill training might slow down the progression of Parkinson’s disease.
1. Introduction
2. Treadmill Exercise on Neural Mitochondrial Functions in Parkinson’s Disease
2.1. Effects of TE Training on Neural Mitochondrial Respiratory Deficiency in PD
2.2. Effects of TE Training on Neural Mitochondrial Biogenesis in PD
2.3. Effects of TE Training on Neural Mitochondrial Dynamics in PD
2.4. Effects of TE Training on Neural Mitophagy in PD
3. Summary
(1) Treadmill training attenuated neural mitochondrial respiratory deficiency in Parkinson’s disease, supported by the evidence that treadmill training normalized the levels of complexes I–V, cytochrome c, and ATP production in the Parkinsonian brain. (2) Treadmill training optimized neural mitochondrial biogenesis in Parkinson’s disease, supported by the evidence that treadmill training increased or normalized the levels of biogenesis regulators (SIRT3, SIRT1, AMPK, PGC-1α, NRF-1,2, and TFAM) and import machinery (TOM-20, TOM-40, TIM-23, and mtHSP70) in the Parkinsonian brain. (3) Treadmill training enhanced the neural mitochondrial fusion in Parkinson’s disease, supported by the evidence that treadmill training increased mitochondrial fusion factors (OPA-1 and MFN-2) in the Parkinsonian brain. (4) Treadmill training repaired the impairment of mitophagy in Parkinson’s disease, supported by the evidence that treadmill training reduced the levels of dysfunctional mitochondria detectors (PINK1, parkin, and p62) and increased the levels of lysosomal factors (LAMP2 and cathepsin L) in the Parkinsonian brain. Taking these findings with the previously hypothesized pathophysiology of Parkinson’s disease together, we drew a hypothesized figure (Figure 1), which suggests that treadmill training could counteract the neurodegeneration of Parkinson’s disease in both the neural mitochondrial respiratory system and neural mitochondrial quality-control.
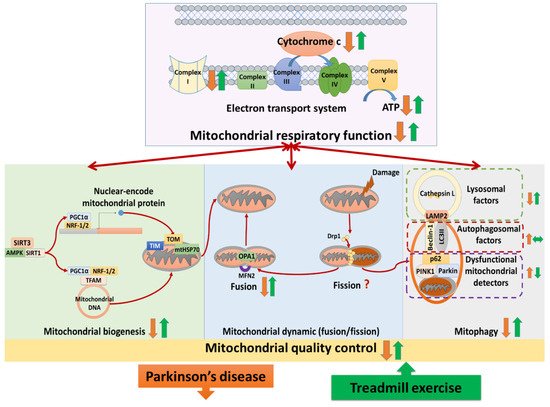
4. The Implications for Future Research
Further interdisciplinary studies are required to investigate the effects of treadmill training on the neural mitochondrial respiratory system, biogenesis, dynamics, and mitophagy in both genetic models and toxin models of Parkinson’s disease. Additionally, clinical studies should clarify the possible therapeutic applications through different exercise interventions into neural mitochondrial dysfunction in Parkinson’s disease.
References
- Sveinbjornsdottir, S. The clinical symptoms of Parkinson’s disease. J. Neurochem. 2016, 139, 318–324.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Cacabelos, R. Parkinson’s disease: From pathogenesis to pharmacogenomics. Int. J. Mol. Sci. 2017, 18, 551.
- Chen, C.; Turnbull, D.M.; Reeve, A.K. Mitochondrial dysfunction in Parkinson’s disease—Cause or consequence? Biology 2019, 8, 38.
- Dossi, G.; Squarcina, L.; Rango, M. In Vivo Mitochondrial Function in Idiopathic and Genetic Parkinson’s Disease. Metabolites 2020, 10, 19.
- Moon, H.E.; Paek, S.H. Mitochondrial dysfunction in Parkinson’s disease. Exp. Neurobiol. 2015, 24, 103–116.
- Banerjee, R.; Starkov, A.A.; Beal, M.F.; Thomas, B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim. Biophys. Acta 2009, 1792, 651–663.
- Meng, H.; Yan, W.-Y.; Lei, Y.-H.; Wan, Z.; Hou, Y.-Y.; Sun, L.-K.; Zhou, J.-P. SIRT3 regulation of mitochondrial quality control in neurodegenerative diseases. Front. Aging Neurosci. 2019, 11, 313.
- Diaz, F.; Moraes, C.T. Mitochondrial biogenesis and turnover. Cell Calcium. 2008, 44, 24–35.
- Lodish, H.; Berk, A.; Kaiser, C.A.; Krieger, M.; Scott, M.P.; Bretscher, A.; Ploegh, H.; Matsudaira, P. Molecular Cell Biology, 6th ed.; W. H. Freeman and Company: New York, NY, USA, 2008.
- Perier, C.; Vila, M. Mitochondrial biology and Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 4, a009332.
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s disease: From pathogenesis to treatment. Cells 2019, 8, 712.
- Mehrholz, J.; Kugler, J.; Storch, A.; Pohl, M.; Elsner, B.; Hirsch, K. Treadmill Training for Patients with Parkinson’s Disease; John Wiley & Sons: Hoboken, NJ, USA, 2015.
- Wang, R.; Tian, H.; Guo, D.; Tian, Q.; Yao, T.; Kong, X. Impacts of exercise intervention on various diseases in rats. J. Sport Health Sci. 2020, 9, 211–227.
- Chuang, C.-S.; Chang, J.-C.; Cheng, F.-C.; Liu, K.-H.; Suc, H.-L.; Liu, C.-S. Modulation of mitochondrial dynamics by treadmill training to improve gait and mitochondrial deficiency in a rat model of Parkinson’s disease. Life Sci. 2017, 191, 236–244.
- Tuon, T.; Souza, P.S.; Santos, M.F.; Pereira, F.T.; Pedroso, G.S.; Luciano, T.F.; Souza, C.T.D.; Dutra, R.C.; Silveira, P.C.L.; Pinho, R.A. Physical training regulates mitochondrial parameters and neuroinflammatory mechanisms in an experimental model of Parkinson’s disease. Oxidative Med. Cell. Longev. 2015, 2015, 261809.
- Jang, Y.; Kwon, I.; Song, W.; Cosio-Lima, L.M.; Taylor, S.; Lee, Y. Modulation of mitochondrial phenotypes by endurance exercise contributes to neuroprotection against a MPTP-induced animal model of PD. Life Sci. 2018, 209, 455–465.
- Ferreira, A.F.F.; Binda, K.H.; Singulani, M.P.; Pereira, C.P.M.; Ferrari, G.D.; Alberici, L.C.; Real, C.C.; Britto, L.R. Physical exercise protects against mitochondria alterations in the 6-hidroxydopamine rat model of Parkinson’s disease. Behav. Brain Res. 2020, 387, 11260.
- Koo, J.-H.; Cho, J.-Y. Treadmill exercise attenuates α-synuclein levels by promoting mitochondrial function and autophagy possibly via SIRT1 in the chronic MPTP/P-induced mouse model of Parkinson’s disease. Neurotox Res. 2017, 32, 473–486.
- Patki, G.; Lau, Y.-S. Impact of exercise on mitochondrial transcription factor expression and damage in the striatum of a chronic mouse model of Parkinson’s disease. Neurosci. Lett. 2011, 505, 268–272.
- Lau, Y.-S.; Patki, G.; Das-Panja, K.; Le, W.-D.; Ahmad, S.O. Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson’s disease with moderate neurodegeneration. Eur. J. Neurosci. 2011, 33, 1264–1274.
- Koo, J.-H.; Cho, J.-Y.; Lee, U.-B. Treadmill exercise alleviates motor deficits and improves mitochondrial import machinery in an MPTP-induced mouse model of Parkinson’s disease. Exp. Gerontol. 2017, 89, 20–29.
- Rezaee, Z.; Marandi, S.M.; Alaei, H.; Esfarjani, F.; Feyzollahzadeh, S. Effects of preventive treadmill exercise on the recovery of metabolic and mitochondrial factors in the 6-hydroxydopamine rat model of Parkinson’s disease. Neurotox. Res. 2019, 35, 908–917.
- Hwang, D.; Koo, J.; Kwon, K.; Choi, D.; Shin, S.; Jeong, J.; Um, H.; Cho, J. Neuroprotective effect of treadmill exercise possibly via regulation of lysosomal degradation molecules in mice with pharmacologically induced Parkinson’s disease. J. Physiol. Sci. 2018, 68, 707–716.

