 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Anton I. Korbut | + 7378 word(s) | 7378 | 2021-08-01 10:55:18 | | | |
| 2 | Anton I. Korbut | -1 word(s) | 7377 | 2021-08-12 12:41:18 | | | | |
| 3 | Vicky Zhou | -1 word(s) | 7377 | 2021-08-12 12:59:16 | | |
Video Upload Options
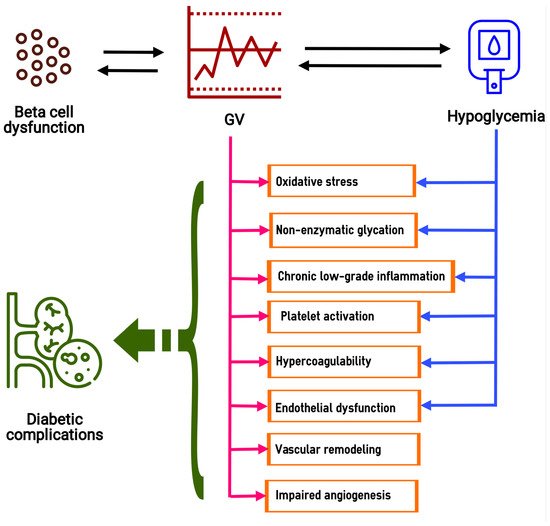
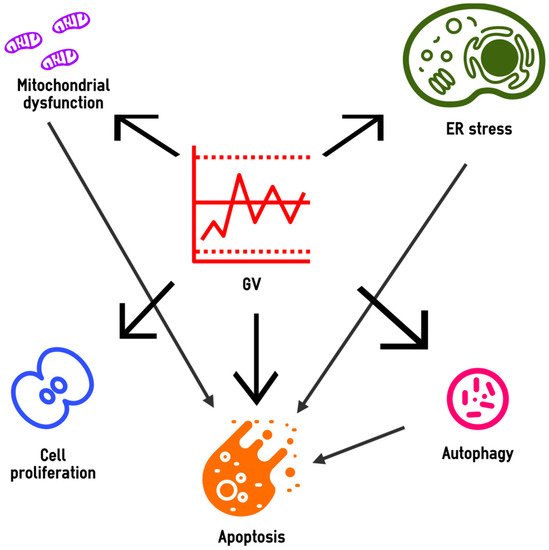
Glucose variability (GV) plays a role in the development of the microvascular and macrovascular complications of diabetes. Current data indicate that the deteriorating effect of GV on target organs can be realized through oxidative stress, glycation, chronic low-grade inflammation, endothelial dysfunction, platelet activation, impaired angiogenesis and renal fibrosis. The effects of GV on oxidative stress, inflammation, endothelial dysfunction and hypercoagulability could be aggravated by hypoglycemia, associated with high GV. Oscillating hyperglycemia contributes to beta cell dysfunction, which leads to a further increase in GV and completes the vicious circle. In cells, the GV-induced cytotoxic effect includes mitochondrial dysfunction, endoplasmic reticulum stress and disturbances in autophagic flux, which are accompanied by reduced viability, activation of apoptosis and abnormalities in cell proliferation. These effects are realized through the up- and down-regulation of a large number of genes and the activity of signaling pathways such as PI3K/Akt, NF-κB, MAPK (ERK), JNK and TGF-β/Smad. Epigenetic modifications mediate the postponed effects of glucose fluctuations. The multiple deteriorative effects of GV provide further support for considering it as a therapeutic target in diabetes.
1. Introduction
2. Biochemical and Pathophysiological Abnormalities Induced by Excessive Glucose Fluctuations
2.1. Oxidative Stress and Non-Enzymatic Glycation
2.2. Chronic Low-Grade Inflammation
2.3. Endothelial Dysfunction and Vascular Remodeling
2.4. Platelet Activation and Hypercoagulability
2.5. Impaired Angiogenesis
2.6. Renal Fibrosis
2.7. Beta Cell Dysfunction
3. Cell Biology under High-GV Conditions
3.1. Altered Mitochondrial Homeostasis
3.2. Endoplasmic Reticulum Stress
3.3. Autophagy
3.4. Apoptosis
3.5. Cell Proliferation
4. Molecular Mechanisms of the High GV Effects in the Target Cells
4.1. Gene Expression
4.2. Epigenetic Modifications
4.3. Signaling Pathways
| Effect | Pathways | Refs. |
|---|---|---|
| Oxidative stress in endothelial and neural cells | PKC/NF-κB, PI3K/Akt, p38MAPK | [99][134][174][175] |
| Endothelial dysfunction and apoptosis | PI3K/Akt, NF-κB, PKC/JNK | [19][58][129][176][184] |
| Proliferation of VSMCs | MAPK (ERK1/2), PI3K/Akt, NF-κB | [138] |
| Vascular low-grade inflammation | NF-κB and p38 MAPK | [162] |
| Renal fibrosis | MAPK (ERK1/2) and TGF- β/Smad | [188] |
| Aortic fibrosis | TGF-β/Smad, NF-κB, p38 MAPK and Runx2 | [185] |
| Neuronal apoptosis and neurodegeneration | PI3K/Akt, NF-κB | [133][134] |
5. Conclusions
Abbreviations
| AGEs | Advanced glycation end-products |
| AMPK | AMP-activated protein kinase |
| ATF4 | Activating transcription factor 4 |
| BMP | Bone morphogenetic protein |
| CARM1 | Coactivator-associated arginine methyltransferase 1 |
| CGM | Continuous glucose monitoring |
| CHOP | C/EBP homologous protein |
| CHG | Constantly high glucose |
| DNMT3b | DNA (cytosine-5-)-methyltransferase 3 beta |
| eNOS | Endothelial nitric oxide synthase |
| EPCs | Endothelial progenitor cells |
| ER | Endoplasmic reticulum |
| ERK | Extracellular signal-regulated kinase |
| GLUT4 | Glucose transporter type 4 |
| GSK3β | Glycogen synthase kinase 3 beta |
| GV | Glucose variability |
| HbA1c | Glycated hemoglobin A1c |
| hsCRP | High-sensitivity C-reactive protein |
| HMGB1 | High-mobility group box 1 |
| HUVECs | Human umbilical vein endothelial cells |
| ICAM-1 | Intercellular adhesion molecules 1 |
| IHG | Intermittently high glucose |
| INS-1 | Insulinoma cells |
| IRF5 | Interferon regulatory factor 5 |
| JNK | c-Jun N-terminal kinase |
| LAMP | Lysosomal-associated membrane protein |
| LC3 | Microtubule-associated proteins 1A/1B light chain 3B |
| MAGE | Mean amplitude of glucose excursions |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein 1 |
| miRNAs | small single-stranded non-coding RNAs |
| mTOR | Mechanistic target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kB |
| PAI-1 | Plasminogen activator inhibitor-1 |
| PI3K | Phosphoinositide-3-kinase |
| PKC | Protein kinase C |
| ROS | Reactive oxygen species |
| RUNX2 | Runt-related transcription factor 2 |
| SHC1 | SHC-transforming protein 1 |
| SKP2 | S-phase kinase-associated protein 2 |
| SD | Standard deviation |
| sICAM-1 | Soluble intercellular adhesion molecules 1 |
| SIRT1 | Sirtuin 1 |
| SGLT1 | Sodium-glucose cotransporter 1 |
| SOD2 | Superoxide dismutase 2 |
| T1D | Type 1 diabetes |
| T2D | Type 2 diabetes |
| TGF-β1 | Transforming growth factor beta 1 |
| TLR4 | Toll-like receptor 4 |
| TNF-α | Tumor necrosis factor α |
| TSP-1 | Thrombospondin-1 |
| TUNEL | Terminal deoxynucleotidyl transferase dUTP nick end labeling |
| VCAM-1 | Vascular cell adhesion molecules 1 |
| VEGF | Vascular endothelial growth factor |
| VSMCs | Vascular smooth muscle cells |
Funding
References
- David Rodbard; Glucose Variability: A Review of Clinical Applications and Research Developments. Diabetes Technology & Therapeutics 2018, 20, S2-5, 10.1089/dia.2018.0092.
- Guillermo E. Umpierrez; Boris P. Kovatchev; Glycemic Variability: How to Measure and Its Clinical Implication for Type 2 Diabetes. The American Journal of the Medical Sciences 2018, 356, 518-527, 10.1016/j.amjms.2018.09.010.
- Thomas Danne; Revital Nimri; Tadej Battelino; Richard M. Bergenstal; Kelly L. Close; J. Hans Devries; Satish Garg; Lutz Heinemann; Irl Hirsch; Stephanie A. Amiel; et al.Roy BeckEmanuele BosiBruce BuckinghamClaudio CobelliEyal DassauFrancis J. DoyleSimon HellerRoman HovorkaWeiping JiaTim JonesOlga KordonouriBoris KovatchevAaron KowalskiLori LaffelDavid MaahsHelen R. MurphyKirsten NorgaardChristopher G. ParkinEric RenardBanshi SabooMauro ScharfWilliam V. TamborlaneStuart A. WeinzimerMoshe Phillip International Consensus on Use of Continuous Glucose Monitoring. Diabetes Care 2017, 40, 1631-1640, 10.2337/dc17-1600.
- Antonio Ceriello; Glucose Variability and Diabetic Complications: Is It Time to Treat?. Diabetes Care 2020, 43, 1169-1171, 10.2337/dci20-0012.
- Zheng Zhou; Bao Sun; Shiqiong Huang; Chunsheng Zhu; Meng Bian; Glycemic variability: adverse clinical outcomes and how to improve it?. Cardiovascular Diabetology 2020, 19, 1-14, 10.1186/s12933-020-01085-6.
- J. Waden; C. Forsblom; L. M. Thorn; D. Gordin; M. Saraheimo; P.-H. Groop; on behalf of the Finnish Diabetic Nephropathy Study Group; A1C Variability Predicts Incident Cardiovascular Events, Microalbuminuria, and Overt Diabetic Nephropathy in Patients With Type 1 Diabetes. Diabetes 2009, 58, 2649-2655, 10.2337/db09-0693.
- Andrea O. Y. Luk; Ronald Ma; Eric Sh Lau; Xilin Yang; Winnie W. Y. Lau; Linda W. L. Yu; Francis C. C. Chow; Juliana Chan; Wing-Yee So; Risk association of HbA1cvariability with chronic kidney disease and cardiovascular disease in type 2 diabetes: prospective analysis of the Hong Kong Diabetes Registry. Diabetes/Metabolism Research and Reviews 2013, 29, 384-390, 10.1002/dmrr.2404.
- Suhua Li; Xixiang Tang; Yanting Luo; Bingyuan Wu; Zhuoshan Huang; Zexiong Li; Long Peng; Yesheng Ling; Jieming Zhu; Junlin Zhong; et al.Jinlai LiuYanming Chen Impact of long-term glucose variability on coronary atherosclerosis progression in patients with type 2 diabetes: a 2.3 year follow-up study. Cardiovascular Diabetology 2020, 19, 1-13, 10.1186/s12933-020-01126-0.
- Yoichiro Hirakawa; Hisatomi Arima; Sophia Zoungas; Toshiharu Ninomiya; Mark Cooper; Pavel Hamet; Giuseppe Mancia; Neil Poulter; Stephen Harrap; Mark Woodward; et al.John Chalmers Impact of Visit-to-Visit Glycemic Variability on the Risks of Macrovascular and Microvascular Events and All-Cause Mortality in Type 2 Diabetes: The ADVANCE Trial. Diabetes Care 2014, 37, 2359-2365, 10.2337/dc14-0199.
- Bernard Zinman; on behalf of the DEVOTE Study Group; Steven P. Marso; Neil R. Poulter; Scott S. Emerson; Thomas R. Pieber; Richard E. Pratley; Martin Lange; Kirstine Brown-Frandsen; Alan Moses; et al.Ann Marie Ocampo FranciscoJesper Barner LekdorfKajsa KvistJohn Buse Day-to-day fasting glycaemic variability in DEVOTE: associations with severe hypoglycaemia and cardiovascular outcomes (DEVOTE 2). Diabetologia 2017, 61, 48-57, 10.1007/s00125-017-4423-z.
- Jin J. Zhou; Dawn C. Schwenke; Gideon Bahn; Peter Reaven; Glycemic Variation and Cardiovascular Risk in the Veterans Affairs Diabetes Trial. Diabetes Care 2018, 41, 2187-2194, 10.2337/dc18-0548.
- Arnaud D Kaze; Prasanna Santhanam; Sebhat Erqou; Rexford S Ahima; Justin Basile Echouffo-Tcheugui; Long-term variability of glycemic markers and risk of all-cause mortality in type 2 diabetes: the Look AHEAD study. BMJ Open Diabetes Research & Care 2020, 8, e001753, 10.1136/bmjdrc-2020-001753.
- Qin-Min Ge; Yan Dong; Hong-Mei Zhang; Qing Su; Effects of intermittent high glucose on oxidative stress in endothelial cells. International Journal of Earth Sciences 2009, 47, 97-103, 10.1007/s00592-009-0140-5.
- Lian-Qing Sun; Bing Xue; Xiao-Jin Li; Xuan Wang; Ling Qu; Ting-Ting Zhang; Jue Zhao; Bao-An Wang; Xiao-Man Zou; Yi-Ming Mu; et al.Ju-Ming Lu Inhibitory effects of Salvianolic acid B on apoptosis of Schwann cells and its mechanism induced by intermittent high glucose. Life Sciences 2012, 90, 99-108, 10.1016/j.lfs.2011.10.001.
- Lian-Qing Sun; Ying-Ying Chen; Xuan Wang; Xiao-Jin Li; Bing Xue; Ling Qu; Ting-Ting Zhang; Yi-Ming Mu; Ju-Ming Lu; The protective effect of Alpha lipoic acid on Schwann cells exposed to constant or intermittent high glucose. Biochemical Pharmacology 2012, 84, 961-973, 10.1016/j.bcp.2012.07.005.
- Jiazhong Sun; Yancheng Xu; Haohua Deng; Suxin Sun; Zhe Dai; Yanlei Sun; Intermittent high glucose exacerbates the aberrant production of adiponectin and resistin through mitochondrial superoxide overproduction in adipocytes. Journal of Molecular Endocrinology 2010, 44, 179-185, 10.1677/jme-09-0088.
- Zhen Zhang; Jing Li; Lei Yang; Rongping Chen; Rui Yang; Hua Zhang; Dehong Cai; Hong Chen; The Cytotoxic Role of Intermittent High Glucose on Apoptosis and Cell Viability in Pancreatic Beta Cells. Journal of Diabetes Research 2014, 2014, 1-9, 10.1155/2014/712781.
- X-G Zhang; Y-Q Zhang; D-K Zhao; J-X Wu; J Zhao; X-M Jiao; B Chen; X-F Lv; Relationship between blood glucose fluctuation and macrovascular endothelial dysfunction in type 2 diabetic patients with coronary heart disease.. European review for medical and pharmacological sciences 2014, 18, 3593-3600.
- Jingshang Wang; Huijun Yin; Ye Huang; Chunyu Guo; Chengdong Xia; Qian Liu; Lu Zhang; Panax Quinquefolius Saponin of Stem and Leaf Attenuates Intermittent High Glucose-Induced Oxidative Stress Injury in Cultured Human Umbilical Vein Endothelial Cells via PI3K/Akt/GSK-3βPathway. Evidence-Based Complementary and Alternative Medicine 2013, 2013, 1-7, 10.1155/2013/196283.
- He Y.-T., Xing S.-S., Gao L., Wang J., Xing Q.-C., Zhang W.; Ginkgo biloba attenuates oxidative DNA damage of human umbilical vein endothelial cells induced by intermittent high glucose. Pharmazie 2014, 69, 203–207.
- Jiazhong SunYancheng XuSuxin Sun; Yancheng Xu; Suxin Sun; Yanlei Sun; Xiang Wang; Intermittent high glucose enhances cell proliferation and VEGF expression in retinal endothelial cells: the role of mitochondrial reactive oxygen species. Molecular and Cellular Biochemistry 2010, 343, 27-35, 10.1007/s11010-010-0495-5.
- Zhangjie Hu; Wenming Fang; Yi Liu; Haowei Liang; Wei Chen; Hui Wang; Acute glucose fluctuation promotes RAGE expression via reactive oxygen species‑mediated NF‑κB activation in rat podocytes. Molecular Medicine Reports 2021, 23, 1-9, 10.3892/mmr.2021.11969.
- Jiazhong Sun; Yancheng Xu; Zhe Dai; Yanlei Sun; Intermittent High Glucose Stimulate MCP-l, IL-18, and PAI-1, but Inhibit Adiponectin Expression and Secretion in Adipocytes Dependent of ROS. Cell Biochemistry and Biophysics 2009, 55, 173-180, 10.1007/s12013-009-9066-3.
- Zhi-Qiang Hou; Hong-Liang Li; Ling Gao; Lin Pan; Jiajun Zhao; Guang-Wei Li; Involvement of chronic stresses in rat islet and INS-1 cell glucotoxicity induced by intermittent high glucose. Molecular and Cellular Endocrinology 2008, 291, 71-78, 10.1016/j.mce.2008.03.004.
- Na Wu; Haitao Shen; Henan Liu; Yanjun Wang; Yu Bai; Ping Han; Acute blood glucose fluctuation enhances rat aorta endothelial cell apoptosis, oxidative stress and pro-inflammatory cytokine expression in vivo. Cardiovascular Diabetology 2016, 15, 1-13, 10.1186/s12933-016-0427-0.
- Chih-Min Chang; Ching-Jung Hsieh; Ju-Chun Huang; I-Chin Huang; Acute and chronic fluctuations in blood glucose levels can increase oxidative stress in type 2 diabetes mellitus. Acta Diabetologica 2012, 49, 171-177, 10.1007/s00592-012-0398-x.
- I. M. E. Wentholt; W. Kulik; R. P. J. Michels; J. B. L. Hoekstra; J. Hans DeVries; Glucose fluctuations and activation of oxidative stress in patients with type 1 diabetes. Diabetologia 2007, 51, 183-190, 10.1007/s00125-007-0842-6.
- Sarah E. Siegelaar; Temo Barwari; Wim Kulik; Joost B. Hoekstra; J. Hans Devries; No Relevant Relationship between Glucose Variability and Oxidative Stress in Well-Regulated Type 2 Diabetes Patients. Journal of Diabetes Science and Technology 2011, 5, 86-92, 10.1177/193229681100500112.
- Yo Kohata; Makoto Ohara; Hiroe Nagaike; Tomoki Fujikawa; Naoya Osaka; Satoshi Goto; Ayako Fukase; Hideki Kushima; Munenori Hiromura; Michishige Terasaki; et al.Yusaku MoriTomoyasu FukuiMotoshi OuchiTatsuya SuzukiTsutomu HiranoSho-Ichi Yamagishi Association of Hemoglobin A1c, 1,5-Anhydro-d-Glucitol and Glycated Albumin with Oxidative Stress in Type 2 Diabetes Mellitus Patients: A Cross-Sectional Study. Diabetes Therapy 2020, 11, 655-665, 10.1007/s13300-020-00772-7.
- Vadim V. Klimontov; Natalia E. Myakina; Glucose variability indices predict the episodes of nocturnal hypoglycemia in elderly type 2 diabetic patients treated with insulin. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2017, 11, 119-124, 10.1016/j.dsx.2016.08.023.
- Ana María Gómez; Diana Cristina Henao; Angélica Imitola Madero; Lucía B. Taboada; Viviana Cruz; María Alejandra Robledo Gómez; Martin Rondón; Oscar Muñoz-Velandia; Maira García-Jaramillo; Fabian León Vargas; et al. Defining High Glycemic Variability in Type 1 Diabetes: Comparison of Multiple Indexes to Identify Patients at Risk of Hypoglycemia. Diabetes Technology & Therapeutics 2019, 21, 430-439, 10.1089/dia.2019.0075.
- Antonio Ceriello; Anna Novials; Emilio Ortega; Lucia La Sala; Gemma Pujadas; Roberto Testa; Anna Rita Bonfigli; Katherine Esposito; Dario Giugliano; Evidence That Hyperglycemia After Recovery From Hypoglycemia Worsens Endothelial Function and Increases Oxidative Stress and Inflammation in Healthy Control Subjects and Subjects With Type 1 Diabetes. Diabetes 2012, 61, 2993-2997, 10.2337/db12-0224.
- Antonio Ceriello; A. Novials; E. Ortega; Gemma Pujadas; L. La Sala; R. Testa; Anna Rita Bonfigli; S. Genovese; Hyperglycemia following recovery from hypoglycemia worsens endothelial damage and thrombosis activation in type 1 diabetes and in healthy controls. Nutrition, Metabolism and Cardiovascular Diseases 2013, 24, 116-123, 10.1016/j.numecd.2013.05.003.
- Sarah Louise Fishman; Halis Sonmez; Craig Basman; Varinder Singh; Leonid Poretsky; The role of advanced glycation end-products in the development of coronary artery disease in patients with and without diabetes mellitus: a review. Molecular Medicine 2018, 24, 1-12, 10.1186/s10020-018-0060-3.
- Roberto Testa; Anna Rita Bonfigli; Francesco Prattichizzo; Lucia La Sala; Valeria De Nigris; Antonio Ceriello; The “Metabolic Memory” Theory and the Early Treatment of Hyperglycemia in Prevention of Diabetic Complications. Nutrients 2017, 9, 437, 10.3390/nu9050437.
- Sho-Ichi Yamagishi; Nobutaka Nakamura; Takanori Matsui; Glycation and cardiovascular disease in diabetes: A perspective on the concept of metabolic memory. Journal of Diabetes 2016, 9, 141-148, 10.1111/1753-0407.12475.
- B. Schisano; G. Tripathi; K. McGee; P. G. McTernan; A. Ceriello; Glucose oscillations, more than constant high glucose, induce p53 activation and a metabolic memory in human endothelial cells. Diabetologia 2011, 54, 1219-1226, 10.1007/s00125-011-2049-0.
- Maddalena Corgnali; Ludovica Piconi; Michael Ihnat; Antonio Ceriello; Evaluation of gliclazide ability to attenuate the hyperglycaemic ‘memory’ induced by high glucose in isolated human endothelial cells. Diabetes/Metabolism Research and Reviews 2007, 24, 301-309, 10.1002/dmrr.804.
- Caroline Pereira Domingueti; Luci Maria Sant'ana Dusse; Maria Das Graças Carvalho; Lirlândia Sousa; Karina Braga Gomes; Ana Paula Fernandes; Diabetes mellitus: The linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. Journal of Diabetes and its Complications 2016, 30, 738-745, 10.1016/j.jdiacomp.2015.12.018.
- Dung Van Nguyen; Lynn C. Shaw; Maria B. Grant; Inflammation in the pathogenesis of microvascular complications in diabetes. Frontiers in Endocrinology 2012, 3, 170, 10.3389/fendo.2012.00170.
- Ludovica Piconi; Lisa Quagliaro; Roberta Assaloni; Roberto Da Ros; Amabile Maier; Gianni Zuodar; Antonio Ceriello; Constant and intermittent high glucose enhances endothelial cell apoptosis through mitochondrial superoxide overproduction. Diabetes/Metabolism Research and Reviews 2006, 22, 198-203, 10.1002/dmrr.613.
- Lisa Quagliaro; Ludovica Piconi; Roberta Assaloni; Roberto Da Ros; Amabile Maier; Gianni Zuodar; Antonio Ceriello; Intermittent high glucose enhances ICAM-1, VCAM-1 and E-selectin expression in human umbilical vein endothelial cells in culture: The distinct role of protein kinase C and mitochondrial superoxide production. Atherosclerosis 2005, 183, 259-267, 10.1016/j.atherosclerosis.2005.03.015.
- Yang Li-Bo; Qi Wen-Bo; Lv Xiao-Hong; Feng You-Lun; Zhang Tie; Intermittent high glucose promotes expression of proinflammatory cytokines in monocytes. Inflammation Research 2010, 60, 367-370, 10.1007/s00011-010-0279-0.
- Chenchen Zhang; Yaxin Bi; Guoxi Jin; Huaiyong Gan; Lei Yu; High and fluctuating glucose levels increase the expression and secretion of interleukin-18 in mouse peritoneal macrophages. Molecular Medicine Reports 2015, 12, 2715-2720, 10.3892/mmr.2015.3753.
- Fatema Al-Rashed; Sardar Sindhu; Hossein Arefanian; Ashraf Al Madhoun; Shihab Kochumon; Reeby Thomas; Sarah Al-Kandari; Abdulwahab Alghaith; Texy Jacob; Fahd Al-Mulla; et al.Rasheed Ahmad Repetitive Intermittent Hyperglycemia Drives the M1 Polarization and Inflammatory Responses in THP-1 Macrophages Through the Mechanism Involving the TLR4-IRF5 Pathway. Cells 2020, 9, 1892, 10.3390/cells9081892.
- Jacqueline M. Ratter; Hanne M.M. Rooijackers; Cees J. Tack; Anneke G.M. Hijmans; Mihai G. Netea; Bastiaan E. De Galan; Rinke Stienstra; Proinflammatory Effects of Hypoglycemia in Humans With or Without Diabetes. Diabetes 2017, 66, 1052-1061, 10.2337/db16-1091.
- Jacqueline M. Ratter; Hanne M. M. Rooijackers; Cor W. M. Jacobs; Bastiaan E. De Galan; Cees J. Tack; Rinke Stienstra; Hypoglycaemia induces recruitment of non-classical monocytes and cytotoxic lymphocyte subsets in type 1 diabetes. Diabetologia 2018, 61, 2069-2071, 10.1007/s00125-018-4683-2.
- Antonio Ceriello; Anna Novials; Emilio Ortega; Silvia Canivell; Lucia La Sala; Gemma Pujadas; Katherine Esposito; Dario Giugliano; Stefano Genovese; Glucagon-Like Peptide 1 Reduces Endothelial Dysfunction, Inflammation, and Oxidative Stress Induced by Both Hyperglycemia and Hypoglycemia in Type 1 Diabetes. Diabetes Care 2013, 36, 2346-2350, 10.2337/dc12-2469.
- Antonio Ceriello; Anna Novials; Emilio Ortega; Silvia Canivell; Gemma Pujadas; Lucia La Sala; Loredana Bucciarelli; Maurizio Rondinelli; Stefano Genovese; Vitamin C further improves the protective effect of GLP-1 on the ischemia-reperfusion-like effect induced by hyperglycemia post-hypoglycemia in type 1 diabetes. Cardiovascular Diabetology 2013, 12, 97-97, 10.1186/1475-2840-12-97.
- Beata Kieć-Wilk; Bartlomiej Matejko; Urszula Razny; Magdalena Stankiewicz; Jan Skupien; Tomasz Klupa; Maciej T. Malecki; Hypoglycemic episodes are associated with inflammatory status in patients with type 1 diabetes mellitus. Atherosclerosis 2016, 251, 334-338, 10.1016/j.atherosclerosis.2016.05.002.
- David Z.I. Cherney; James W. Scholey; Etienne Sochett; Timothy J. Bradley; Heather N. Reich; The Acute Effect of Clamped Hyperglycemia on the Urinary Excretion of Inflammatory Cytokines/Chemokines in Uncomplicated Type 1 Diabetes: A pilot study. Diabetes Care 2010, 34, 177-180, 10.2337/dc10-1219.
- Eik W., Marcon S.S., Krupek T., Previdelli I.T.S., Pereira O.C.N., Silva M.a.R.C.P., Bazotte R.B.; Blood levels of pro-inflammatory and anti-inflammatory cytokines during an oral glucose tolerance test in patients with symptoms suggesting reactive hypoglycemia. Braz. J. Med. Biol. Res. 2016, 49, e5195.
- Juliana B. Drummond; Izabela Barbosa; Robert Dantzer; Antonio Lucio Teixeira; The effect of insulin-induced hypoglycemia on inflammatory markers: A systematic review. Brain, Behavior, and Immunity 2018, 73, 41-50, 10.1016/j.bbi.2018.05.003.
- Abdurrahman Kaya; Cemil Koçyiğit; Gönül Çatlı; Elif Büşra Özkan; Bumin Nuri Dündar; The Relationship Between Glycemic Variability and Inflammatory Markers in Obese Children with Insulin Resistance and Metabolic Syndrome. Journal of Clinical Research in Pediatric Endocrinology 2017, 9, 202-207, 10.4274/jcrpe.4031.
- Vadim Klimontov; N V Tyan; O N Fazullina; N E Myakina; N B Orlov; V I Konenkov; Acute-phase serum proteins and adipocytokines in women with type 2 diabetes mellitus: Relationships with body composition and blood glucose fluctuations. Terapevticheskii arkhiv 2016, 88, 35-41, 10.17116/terarkh2016881035-41.
- Silvio Buscemi; Salvatore Verga; Santina Cottone; Vitalba Azzolina; Barbara Buscemi; Diana Gioia; Giovanni Cerasola; Glycaemic variability and inflammation in subjects with metabolic syndrome. International Journal of Earth Sciences 2008, 46, 55-61, 10.1007/s00592-008-0061-8.
- Shalini Paul; Azam Ali; Rajesh Katare; Molecular complexities underlying the vascular complications of diabetes mellitus – A comprehensive review. Journal of Diabetes and its Complications 2020, 34, 107613, 10.1016/j.jdiacomp.2020.107613.
- Jie Liao; Minxiang Lei; Xiong Chen; Feng Liu; Effect of intermittent high glucose on synthesis of nitric oxide in human umbilical vein endothelial cells and its mechanism.. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2010, 35, 295–300, 10.3969/j.issn.1672-7347.2010.04.003.
- Katarína Kuricová; Lukas Pacal; Jan Soupal; Martin Prázný; Katerina Kankova; Effect of glucose variability on pathways associated with glucotoxicity in diabetes: Evaluation of a novel in vitro experimental approach. Diabetes Research and Clinical Practice 2016, 114, 1-8, 10.1016/j.diabres.2016.02.006.
- Morihiko Maeda; Toshio Hayashi; Natsumi Mizuno; Yuichi Hattori; Masafumi Kuzuya; Intermittent High Glucose Implements Stress-Induced Senescence in Human Vascular Endothelial Cells: Role of Superoxide Production by NADPH Oxidase. PLOS ONE 2015, 10, e0123169, 10.1371/journal.pone.0123169.
- Eszter Maria Horvath; R. Benkő; L. Kiss; M. Murányi; T. Pék; K. Fekete; Tamas Barany; Á. Somlai; A. Csordás; C. Szabo; et al. Rapid ‘glycaemic swings’ induce nitrosative stress, activate poly(ADP-ribose) polymerase and impair endothelial function in a rat model of diabetes mellitus. Diabetologia 2009, 52, 952-961, 10.1007/s00125-009-1304-0.
- Alessandra Di Flaviani; Fabiana Picconi; Paola Di Stefano; Ilaria Giordani; Ilaria Malandrucco; Paola Maggio; Paola Palazzo; Fabrizio Sgreccia; Carlo Peraldo; Fabrizio Farina; et al.Gaetano FrajeseSimona Frontoni Impact of Glycemic and Blood Pressure Variability on Surrogate Measures of Cardiovascular Outcomes in Type 2 Diabetic Patients. Diabetes Care 2011, 34, 1605-1609, 10.2337/dc11-0034.
- Alexia S. Peña; Jennifer J. Couper; Jennifer Harrington; Roger Gent; Jan Fairchild; Elaine Tham; Peter Baghurst; Hypoglycemia, but Not Glucose Variability, Relates to Vascular Function in Children with Type 1 Diabetes. Diabetes Technology & Therapeutics 2012, 14, 457-462, 10.1089/dia.2011.0229.
- Ahmed Al-Qaissi; Maria Papageorgiou; Harshal Deshmukh; Leigh A. Madden; Alan Rigby; Eric S. Kilpatrick; Stephen L. Atkin; Thozhukat Sathyapalan; Effects of acute insulin-induced hypoglycaemia on endothelial microparticles in adults with and without type 2 diabetes. Diabetes, Obesity and Metabolism 2018, 21, 533-540, 10.1111/dom.13548.
- Bedile Irem Tiftikcioglu; Sule Bilgin; Tarik Duksal; Sukran Kose; Yasar Zorlu; Autonomic Neuropathy and Endothelial Dysfunction in Patients With Impaired Glucose Tolerance or Type 2 Diabetes Mellitus. Medicine 2016, 95, e3340-e3340, 10.1097/md.0000000000003340.
- Yuri D. Foreman; Martijn C. G. J. Brouwers; Tos Tjm Berendschot; Martien C. J. M. Van Dongen; Simone J. P. M. Eussen; Marleen M. J. Van Greevenbroek; Ronald M. A. Henry; Alfons J. H. M. Houben; Carla J. H. Van Der Kallen; Abraham A. Kroon; et al.Koen D. ReesinkMiranda SchramNicolaas SchaperCoen D. A. Stehouwer The oral glucose tolerance test-derived incremental glucose peak is associated with greater arterial stiffness and maladaptive arterial remodeling: The Maastricht Study. Cardiovascular Diabetology 2019, 18, 1-12, 10.1186/s12933-019-0950-x.
- Li Zhang; Haichen Sun; Shuang Liu; Jinhuan Gao; Jinggang Xia; Glycemic variability is associated with vascular calcification by the markers of endoplasmic reticulum stress-related apoptosis, Wnt1, galectin-3 and BMP-2.. Diabetology & Metabolic Syndrome 2019, 11, 67-8, 10.1186/s13098-019-0464-4.
- Tatiana E. Suslova; Alexei V. Sitozhevskii; Oksana Ogurkova; Elena Kravchenko; Irina V. Kologrivova; Nina D. Anfinogenova; Rostislav Karpov; Platelet hemostasis in patients with metabolic syndrome and type 2 diabetes mellitus: cGMP- and NO-dependent mechanisms in the insulin-mediated platelet aggregation. Frontiers in Physiology 2015, 5, 501, 10.3389/fphys.2014.00501.
- F. Santilli; G. Formoso; P. Sbraccia; Maurizio Averna; R. Miccoli; P. Di Fulvio; A. Ganci; N. Pulizzi; S. Lattanzio; G. Ciabattoni; et al.A. ConsoliR. LauroC. PatronoGiovanni Davi Postprandial hyperglycemia is a determinant of platelet activation in early type 2 diabetes mellitus. Journal of Thrombosis and Haemostasis 2010, 8, 828-837, 10.1111/j.1538-7836.2010.03742.x.
- Annunziata Nusca; Dario Tuccinardi; Claudio Proscia; Rosetta Melfi; Silvia Manfrini; Antonio Nicolucci; Antonio Ceriello; Paolo Pozzilli; Gian Paolo Ussia; Francesco Grigioni; et al.Germano Di Sciascio Incremental role of glycaemic variability over HbA1c in identifying type 2 diabetic patients with high platelet reactivity undergoing percutaneous coronary intervention. Cardiovascular Diabetology 2019, 18, 1-9, 10.1186/s12933-019-0952-8.
- Guoqiang Gu; Liqiang Chen; Mian Wang; Wei Cui; Acute Fluctuation in Blood Glucose has no Effect on Platelet Aggregation Rate. Clinical Laboratory 2014, 60, 1071-3, 10.7754/clin.lab.2013.130711.
- Ahmed Iqbal; Lynne Prince; Peter Novodvorsky; Alan Bernjak; Mark Thomas; Lewis Birch; Danielle Lambert; Linda J Kay; Fiona J Wright; Ian A Macdonald; et al.Richard JacquesRobert StoreyRory McCrimmonSheila FrancisSimon R HellerIan Sabroe Effect of Hypoglycemia on Inflammatory Responses and the Response to Low-Dose Endotoxemia in Humans. The Journal of Clinical Endocrinology & Metabolism 2018, 104, 1187-1199, 10.1210/jc.2018-01168.
- Paulo Zoé Costa; Raquel Soares; Neovascularization in diabetes and its complications. Unraveling the angiogenic paradox. Life Sciences 2013, 92, 1037-1045, 10.1016/j.lfs.2013.04.001.
- Jun Guo; Yaxiong Sang; Tao Yin; Bo Wang; Wenping Yang; Xue Li; Huan Li; Yan Kang; miR-1273g-3p participates in acute glucose fluctuation-induced autophagy, dysfunction, and proliferation attenuation in human umbilical vein endothelial cells. American Journal of Physiology-Endocrinology and Metabolism 2016, 310, E734-E743, 10.1152/ajpendo.00444.2015.
- Federico Biscetti; Dario Pitocco; Giuseppe Straface; Francesco Zaccardi; Raimondo De Cristofaro; Paola Rizzo; Stefano Lancellotti; Vincenzo Arena; Egidio Stigliano; Tittania Musella; et al.Giovanni GhirlandaAndrea Flex Glycaemic variability affects ischaemia-induced angiogenesis in diabetic mice. Clinical Science 2011, 121, 555-564, 10.1042/cs20110043.
- Lidong Hu; Si-Cheng Dai; Xiaojun Luan; Jingsong Chen; Anthony Cannavicci; Dysfunction and Therapeutic Potential of Endothelial Progenitor Cells in Diabetes Mellitus. Journal of Clinical Medicine Research 2018, 10, 752-757, 10.14740/jocmr3581w.
- Yuiko Inaba; Chiharu Tsutsumi; Fumitaka Haseda; Reiko Fujisawa; Shinobu Mitsui; Hiroyuki Sano; JungO Terasaki; Toshiaki Hanafusa; Akihisa Imagawa; Impact of glycemic variability on the levels of endothelial progenitor cells in patients with type 1 diabetes. Diabetology International 2017, 9, 113-120, 10.1007/s13340-017-0335-x.
- Maria Ida Maiorino; Ofelia Casciano; Elisabetta Della Volpe; Giuseppe Bellastella; Dario Giugliano; Katherine Esposito; Reducing glucose variability with continuous subcutaneous insulin infusion increases endothelial progenitor cells in type 1 diabetes: an observational study. Endocrine 2015, 52, 244-252, 10.1007/s12020-015-0686-7.
- F. Boscari; M. D’Anna; B. M. Bonora; S. Tresso; R. Cappellari; A. Avogaro; D. Bruttomesso; G. P. Fadini; Effects of glucose variability on hematopoietic stem/progenitor cells in patients with type 1 diabetes. Journal of Endocrinological Investigation 2020, 44, 119-126, 10.1007/s40618-020-01278-6.
- A. Goth; L. Lengyel; N. Nådasdi; C. Sávely; Renal Lesions due to Fluctuations in Blood Sugar Levels. Acta Medica Scandinavica 2009, 158, 475-480, 10.1111/j.0954-6820.1957.tb15514.x.
- A. Takeuchi; D. C. Throckmorton; A. P. Brogden; N. Yoshizawa; H. Rasmussen; M. Kashgarian; Periodic high extracellular glucose enhances production of collagens III and IV by mesangial cells. American Journal of Physiology-Renal Physiology 1995, 268, F13-F19, 10.1152/ajprenal.1995.268.1.f13.
- S. C. Jones; H. J. Saunders; W. Qi; C. A. Pollock; Intermittent high glucose enhances cell growth and collagen synthesis in cultured human tubulointerstitial cells. Diabetologia 1999, 42, 1113-1119, 10.1007/s001250051279.
- T. S. Polhill; S. Saad; P. Poronnik; G. R. Fulcher; C. A. Pollock; Short-term peaks in glucose promote renal fibrogenesis independently of total glucose exposure. American Journal of Physiology-Renal Physiology 2004, 287, F268-F273, 10.1152/ajprenal.00084.2004.
- Changjiang Ying; Xiaoyan Zhou; Zhenzhen Chang; Hongwei Ling; Xingbo Cheng; Wei Li; Blood glucose fluctuation accelerates renal injury involved to inhibit the AKT signaling pathway in diabetic rats. Endocrine 2016, 53, 81-96, 10.1007/s12020-016-0867-z.
- Ya-Fei Yang; Tsai-Chung Li; Chia-Ing Li; Chiu-Shong Liu; Wen-Yuan Lin; Sing-Yu Yang; Jen-Huai Chiang; Chiu-Ching Huang; Fung-Chang Sung; Cheng-Chieh Lin; et al. Visit-to-Visit Glucose Variability Predicts the Development of End-Stage Renal Disease in Type 2 Diabetes. Medicine 2015, 94, e1804-e1804, 10.1097/md.0000000000001804.
- Akiko Takenouchi; Ayaka Tsuboi; Mayu Terazawa-Watanabe; Miki Kurata; Keisuke Fukuo; Tsutomu Kazumi; Direct association of visit-to-visit HbA1c variation with annual decline in estimated glomerular filtration rate in patients with type 2 diabetes. Journal of Diabetes & Metabolic Disorders 2015, 14, 1-7, 10.1186/s40200-015-0201-y.
- Fraser W. Gibb; John A. McKnight; Catriona Clarke; Mark W. J. Strachan; Preserved C-peptide secretion is associated with fewer low-glucose events and lower glucose variability on flash glucose monitoring in adults with type 1 diabetes. Diabetologia 2020, 63, 906-914, 10.1007/s00125-020-05099-3.
- Merete B. Christensen; Peter Gæde; Eva Hommel; Anders Gotfredsen; Kirsten Nørgaard; Glycaemic variability and hypoglycaemia are associated with C-peptide levels in insulin-treated type 2 diabetes. Diabetes & Metabolism 2019, 46, 61-65, 10.1016/j.diabet.2019.02.002.
- Yusheng Zhang; Jiao Dai; Xiao Han; Yue Zhao; Hui Zhang; Xuan Liu; Wei Li; Hongwei Ling; Xiaoyan Zhou; Changjiang Ying; et al. Glycemic variability indices determined by self-monitoring of blood glucose are associated with β-cell function in Chinese patients with type 2 diabetes. Diabetes Research and Clinical Practice 2020, 164, 108152, 10.1016/j.diabres.2020.108152.
- Ye-Hwang Cheong; Mi-Kyung Kim; Moon-Ho Son; Bong-Kiun Kaang; Glucose exposure pattern determines glucagon-like peptide 1 receptor expression and signaling through endoplasmic reticulum stress in rat insulinoma cells. Biochemical and Biophysical Research Communications 2011, 414, 220-225, 10.1016/j.bbrc.2011.09.061.
- Shao C., Gu J., Meng X., Zheng H., Wang D.; Systematic Investigation into the role of intermittent high glucose in pancreatic beta-cells. Int. J. Clin. Exp. Med. 2015;, 8:, 5462–5469.
- Jun Wada; Atsuko Nakatsuka; Mitochondrial Dynamics and Mitochondrial Dysfunction in Diabetes.. Acta Med. Okayama 2016, 70, 151-8, 10.18926/amo/54413.
- Daniel Galvan; Nathanael H. Green; Farhad R. Danesh; The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney International 2017, 92, 1051-1057, 10.1016/j.kint.2017.05.034.
- Meng-Yu Wu; Giou-Teng Yiang; Tzu-Ting Lai; Chia-Jung Li; The Oxidative Stress and Mitochondrial Dysfunction during the Pathogenesis of Diabetic Retinopathy. Oxidative Medicine and Cellular Longevity 2018, 2018, 1-12, 10.1155/2018/3420187.
- Aleksandra Cieluch; Aleksandra Uruska; Dorota Zozulinska-Ziolkiewicz; Can We Prevent Mitochondrial Dysfunction and Diabetic Cardiomyopathy in Type 1 Diabetes Mellitus? Pathophysiology and Treatment Options. International Journal of Molecular Sciences 2020, 21, 2852, 10.3390/ijms21082852.
- Milagros Rocha; Noelia Diaz-Morales; Susana Rovira-Llopis; Irene Escribano-Lopez; Celia Bañuls; Antonio Hernandez-Mijares; Evanthia Diamanti-Kandarakis; Victor M. Victor; Mitochondrial Dysfunction and Endoplasmic Reticulum Stress in Diabetes. Current Pharmaceutical Design 2016, 22, 2640-2649, 10.2174/1381612822666160209152033.
- Lucia La Sala; Simona Mrakic-Sposta; Stefano Micheloni; Francesco Prattichizzo; Antonio Ceriello; Glucose-sensing microRNA-21 disrupts ROS homeostasis and impairs antioxidant responses in cellular glucose variability. Cardiovascular Diabetology 2018, 17, 1-14, 10.1186/s12933-018-0748-2.
- Shanjun Tao; Younan Ren; Haowen Zheng; Mengqiu Zhao; Xu Zhang; Yuanmei Zhu; Jieren Yang; Shuguo Zheng; Salvianolic acid B inhibits intermittent high glucose-induced INS-1 cell apoptosis through regulation of Bcl-2 proteins and mitochondrial membrane potential. European Journal of Pharmacology 2017, 814, 56-62, 10.1016/j.ejphar.2017.08.007.
- André Quincozes-Santos; Larissa Daniele Bobermin; Adriano de Assis; Carlos-Alberto Gonçalves; Diogo Souza; Fluctuations in glucose levels induce glial toxicity with glutamatergic, oxidative and inflammatory implications. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2017, 1863, 1-14, 10.1016/j.bbadis.2016.09.013.
- Xueyao Yin; Fenping Zheng; Qianqian Pan; Saifei Zhang; Dan Yu; Zhiye Xu; Hong Li; Glucose fluctuation increased hepatocyte apoptosis under lipotoxicity and the involvement of mitochondrial permeability transition opening. Journal of Molecular Endocrinology 2015, 55, 169-181, 10.1530/jme-15-0101.
- Shotaro Saito; Luong Cong Thuc; Yasushi Teshima; Chisato Nakada; Satoru Nishio; Hidekazu Kondo; Akira Fukui; Ichitaro Abe; Yuki Ebata; Tetsunori Saikawa; et al.Masatsugu MoriyamaNaohiko Takahashi Glucose Fluctuations Aggravate Cardiac Susceptibility to Ischemia/Reperfusion Injury by Modulating MicroRNAs Expression. Circulation Journal 2016, 80, 186-195, 10.1253/circj.cj-14-1218.
- Isabelle Chareyron; Stefan Christen; Sofia Moco; Armand Valsesia; Steve Lassueur; Loïc Dayon; Claes B. Wollheim; Jaime Santo Domingo; Andreas Wiederkehr; Augmented mitochondrial energy metabolism is an early response to chronic glucose stress in human pancreatic beta cells. Diabetologia 2020, 63, 2628-2640, 10.1007/s00125-020-05275-5.
- Wei-Long Xu; Su Liu; Nan Li; Li-Fang Ye; Min Zha; Chang-Yin Li; Yue Zhao; Qiang Pu; Jin-Jing Bao; Xing-Jie Chen; et al.Jiang-Yi YuYing-Hao Pei Quercetin Antagonizes Glucose Fluctuation Induced Renal Injury by Inhibiting Aerobic Glycolysis via HIF-1α/miR-210/ISCU/FeS Pathway. Frontiers in Medicine 2021, 8, 656086, 10.3389/fmed.2021.656086.
- Thaia Silva-Rodrigues; Eduardo De-Souza-Ferreira; Caio Mota Machado; Bruno Cabral; Clara Rodrigues-Ferreira; Antonio Galina; Hyperglycemia in a type 1 Diabetes Mellitus model causes a shift in mitochondria coupled-glucose phosphorylation and redox metabolism in rat brain. Free Radical Biology and Medicine 2020, 160, 796-806, 10.1016/j.freeradbiomed.2020.09.017.
- Imke L. Lemmer; Nienke Willemsen; Nazia Hilal; Alexander Bartelt; A guide to understanding endoplasmic reticulum stress in metabolic disorders. Molecular Metabolism 2021, 47, 101169, 10.1016/j.molmet.2021.101169.
- Himanshu Sankrityayan; Manisha J. Oza; Yogesh A. Kulkarni; Shrikant R. Mulay; Anil Bhanudas Gaikwad; ER stress response mediates diabetic microvascular complications. Drug Discovery Today 2019, 24, 2247-2257, 10.1016/j.drudis.2019.08.003.
- Yimin Zhong; Joshua J. Wang; Sarah X. Zhang; Intermittent But Not Constant High Glucose Induces ER Stress and Inflammation in Human Retinal Pericytes. Advances in Experimental Medicine and Biology 2011, 723, 285-292, 10.1007/978-1-4614-0631-0_37.
- Kengo Ikesugi; Michael L. Mulhern; Christian J. Madson; Ken-Ichi Hosoya; Tetsuya Terasaki; Peter F. Kador; Toshimichi Shinohara; Induction of Endoplasmic Reticulum Stress in Retinal Pericytes by Glucose Deprivation. Current Eye Research 2006, 31, 947-953, 10.1080/02713680600966785.
- Ayako Kato; Yasuaki Tatsumi; Hideji Yako; Kazunori Sango; Tatsuhito Himeno; Masaki Kondo; Yoshiro Kato; Hideki Kamiya; Jiro Nakamura; Koichi Kato; et al. Recurrent short-term hypoglycemia and hyperglycemia induce apoptosis and oxidative stress via the ER stress response in immortalized adult mouse Schwann (IMS32) cells. Neuroscience Research 2018, 147, 26-32, 10.1016/j.neures.2018.11.004.
- Andrew T. Sage; Sarah Holtby-Ottenhof; Yuanyuan Shi; Suzana Damjanovic; Arya M. Sharma; Geoff H. Werstuck; Metabolic Syndrome and Acute Hyperglycemia Are Associated With Endoplasmic Reticulum Stress in Human Mononuclear Cells. Obesity 2012, 20, 748-755, 10.1038/oby.2011.144.
- Tadashi Ichimiya; Tsukasa Yamakawa; Takehiro Hirano; Yoshihiro Yokoyama; Yuki Hayashi; Daisuke Hirayama; Kohei Wagatsuma; Takao Itoi; Hiroshi Nakase; Autophagy and Autophagy-Related Diseases: A Review. International Journal of Molecular Sciences 2020, 21, 8974, 10.3390/ijms21238974.
- Noboru Mizushima; Beth Levine; Autophagy in Human Diseases. New England Journal of Medicine 2020, 383, 1564-1576, 10.1056/nejmra2022774.
- Michelle R Marasco; Amelia K Linnemann; β-Cell Autophagy in Diabetes Pathogenesis. Endocrinology 2018, 159, 2127-2141, 10.1210/en.2017-03273.
- Charanya Muralidharan; Abass M. Conteh; Michelle R. Marasco; Justin J. Crowder; Jeroen Kuipers; Pascal de Boer; Amelia K. Linnemann; Pancreatic beta cell autophagy is impaired in type 1 diabetes. Diabetologia 2021, 64, 865-877, 10.1007/s00125-021-05387-6.
- Tao Tao; Huanbai Xu; Autophagy and Obesity and Diabetes. Advances in Experimental Medicine and Biology 2020, 1207, 445-461, 10.1007/978-981-15-4272-5_32.
- Jinyoung Kim; Yu-Mi Lim; Myung-Shik Lee; The Role of Autophagy in Systemic Metabolism and Human-Type Diabetes. Molecules and Cells 2018, 41, 11-17, 10.14348/molcells.2018.2228.
- Tien-An Lin; Victor Chien-Chia Wu; Chao-Yung Wang; Autophagy in Chronic Kidney Diseases. Cells 2019, 8, 61, 10.3390/cells8010061.
- Danyi Yang; Man J. Livingston; Zhiwen Liu; Guie Dong; Ming Zhang; Jian-Kang Chen; Zheng Dong; Autophagy in diabetic kidney disease: regulation, pathological role and therapeutic potential. Cellular and Molecular Life Sciences 2017, 75, 669-688, 10.1007/s00018-017-2639-1.
- Anton I. Korbut; Iuliia S. Taskaeva; Nataliya P. Bgatova; Natalia A. Muraleva; Nikolai B. Orlov; Maksim V. Dashkin; Anna Khotskina; Evgenii L. Zavyalov; Vladimir I. Konenkov; Thomas Klein; et al.Vadim V. Klimontov SGLT2 Inhibitor Empagliflozin and DPP4 Inhibitor Linagliptin Reactivate Glomerular Autophagy in db/db Mice, a Model of Type 2 Diabetes. International Journal of Molecular Sciences 2020, 21, 2987, 10.3390/ijms21082987.
- Alejo Efeyan; William C. Comb; David M. Sabatini; Nutrient-sensing mechanisms and pathways. Nature 2015, 517, 302-310, 10.1038/nature14190.
- Daniel J. Klionsky; Amal Kamal Abdel-Aziz; Sara Abdelfatah; Mahmoud Abdellatif; Asghar Abdoli; Steffen Abel; Hagai Abeliovich; Marie H. Abildgaard; Yakubu Princely Abudu; Abraham Acevedo-Arozena; et al.Iannis E. AdamopoulosKhosrow AdeliTimon E. AdolphAnnagrazia AdornettoElma AflakiGalila AgamAnupam AgarwalBharat B. AggarwalMaria AgnelloPatrizia AgostinisJaved N. AgrewalaAlexander AgrotisPatricia V. AguilarS. Tariq AhmadZubair M. AhmedUlises Ahumada-CastroSonja AitsShu AizawaYunus AkkocTonia AkoumianakiHafize Aysin AkpinarAhmed M. Al-AbdLina Al-AkraAbeer Al-GharaibehMoulay A. Alaoui-JamaliSimon AlbertiElísabet Alcocer-GómezCristiano AlessandriMuhammad AliM. Abdul Alim Al-BariSaeb AliwainiJavad AlizadehEugènia AlmacellasAlexandru AlmasanAlicia AlonsoGuillermo D. AlonsoNihal Altan-BonnetDario C. AltieriÉlida M. C. ÁlvarezSara AlvesCristine Alves da CostaMazen M. AlzaharnaMarialaura AmadioConsuelo AmantiniCristina AmaralSusanna AmbrosioAmal O. AmerVeena AmmanathanZhenyi AnStig U. AndersenShaida A. AndrabiMagaiver Andrade-SilvaAllen M. AndresSabrina AngeliniDavid AnnUche C. AnozieMohammad Y. AnsariPedro AntasAdam AntebiZuriñe AntónTahira AnwarLionel ApetohNadezda ApostolovaToshiyuki ArakiYasuhiro ArakiKohei ArasakiWagner L. AraújoJun ArayaCatherine ArdenMaria-Angeles ArévaloSandro ArguellesEsperanza AriasJyothi ArikkathHirokazu ArimotoAileen R. AriosaDarius Armstrong-JamesLaetitia Arnauné-PelloquinAngeles ArocaDaniela S. ArroyoIvica ArsovRubén ArteroDalia Maria Lucia AsaroMichael AschnerMilad AshrafizadehOsnat Ashur-FabianAtanas G. AtanasovAlicia K. AuPatrick AubergerHolger W. AunerLaure AurelianRiccardo AutelliLaura AvaglianoYenniffer ÁvalosSanja AveicCélia Alexandra AveleiraTamar Avin-WittenbergYucel AydinScott AytonSrinivas AyyadevaraMaria AzzopardiMisuzu BabaJonathan M. BackerSteven K. BackuesDong-Hun BaeOk-Nam BaeSoo Han BaeEric H. BaehreckeAhruem BaekSeung-Hoon BaekSung Hee BaekGiacinto BagettaAgnieszka Bagniewska-ZadwornaHua BaiJie BaiXiyuan BaiYidong BaiNandadulal BairagiShounak BaksiTeresa BalbiCosima T. BaldariWalter BalduiniAndrea BallabioMaria BallesterSalma BalazadehRena BalzanRina BandopadhyaySreeparna BanerjeeSulagna BanerjeeÁgnes BánrétiYan BaoMauricio S. BaptistaAlessandra BaraccaCristiana BarbatiAriadna BargielaDaniela BarilàPeter G. BarlowSami J. BarmadaEsther BarreiroGeorge E. BarretoJiri BartekBonnie BartelAlberto BartolomeGaurav R. BarveSuresh H. BasagoudanavarDiane C. BasshamRobert C. BastAlakananda BasuHenri BatokoIsabella BattenEtienne E. BaulieuBradley L. BaumgarnerJagadeesh BayryRupert BealeIsabelle BeauFlorian BeaumatinLuiz R.G. BecharaGeorge R. BeckMichael F. BeersJakob BegunChristian BehrendsGeorg M.N. BehrensRoberto BeiEloy BejaranoShai BelChristian BehlAmine BelaidNaïma Belgareh-TouzéCristina BellarosaFrancesca BelleudiMelissa Belló PérezRaquel Bello-MoralesJackeline Soares De Oliveira BeltranSebastián BeltranDoris Mangiaracina BenbrookMykolas BendoriusBruno A. BenitezIrene Benito-CuestaJulien BensalemMartin W. BerchtoldSabina BerezowskaDaniele BergamaschiMatteo BergamiAndreas BergmannLaura BerliocchiClarisse Berlioz-TorrentAmélie BernardLionel BerthouxCagri G. BesirliSebastien BesteiroVirginie M. BetinRudi BeyaertJelena S. BezbradicaKiran BhaskarIngrid Bhatia-KissovaResham BhattacharyaSujoy BhattacharyaShalmoli BhattacharyyaShenuarin BhuiyanSujit Kumar BhutiaLanrong BiXiaolin BiTrevor J. BidenKrikor BijianViktor A. BillesNadine BinartClaudia BincolettoAsa B. BirgisdottirGeir BjorkoyGonzalo BlancoAna Blas-GarciaJanusz BlasiakRobert BlomgranKlas BlomgrenJanice S. BlumEmilio Boada-RomeroMirta BobanKathleen Boesze-BattagliaPhilippe BoeufBarry BolandPascale BomontPaolo BonaldoSrinivasa Reddy BonamLaura BonfiliJuan S. BonifacinoBrian A. BooneMartin D. BootmanMatteo BordiChristoph BornerBeat C. BornhauserGautam BorthakurJürgen BoschSantanu BoseLuis M. BotanaJuan BotasChantal M. BoulangerMichael E. BoultonMathieu BourdenxBenjamin BourgeoisNollaig M. BourkeGuilhem BousquetPatricia BoyaPeter V. BozhkovLuiz H. M. BoziTolga O. BozkurtDoug E. BrackneyChristian H. BrandtsRalf J. BraunGerhard H. BrausRoberto Bravo-SaguaJosé M. Bravo-San PedroPatrick BrestMarie-Agnès BringerAlfredo Briones-HerreraV. Courtney BroaddusPeter BrodersenJeffrey L. BrodskySteven L. BrodyPaola G. BronsonJeff M. BronsteinCarolyn N. BrownRhoderick E. BrownPatricia C. BrumJohn H. BrumellNicola Brunetti-PierriDaniele BrunoRobert J. Bryson-RichardsonCecilia BucciCarmen BuchrieserMarta BuenoLaura Elisa Buitrago-MolinaSimone BuraschiShilpa BuchJ. Ross BuchanErin M. BuckinghamHikmet BudakMauricio BudiniGeert BultynckFlorin BuradaJoseph R. BurgoyneM. Isabel BurónVictor BustosSabrina BüttnerElena ButturiniAaron ByrdIsabel CabasSandra Cabr Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382, 10.1080/15548627.2020.1797280.
- Karyn E. King; Truc T. Losier; Ryan C. Russell; Regulation of Autophagy Enzymes by Nutrient Signaling. Trends in Biochemical Sciences 2021, 46(8):, 687-700, 10.1016/j.tibs.2021.01.006.
- Kendall J. Condon; David M. Sabatini; Nutrient regulation of mTORC1 at a glance. Journal of Cell Science 2019, 132, jcs222570, 10.1242/jcs.222570.
- Zhang N., Zhao Y.; Other molecular mechanisms regulating autophagy. Adv. Exp. Med. Biol. 2019;, 1206:, 261–271..
- Safa Abdelgadir Mohamed Elhassan; Mayuren Candasamy; Elaine Wan Ling Chan; Subrat Kumar Bhattamisra; Elaine Chan Wan Ling; Autophagy and GLUT4: The missing pieces. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2018, 12, 1109-1116, 10.1016/j.dsx.2018.05.020.
- Hong Zhao; Mingzhu Tang; Meiqing Liu; Linxi Chen; Glycophagy: An emerging target in pathology. Clinica Chimica Acta 2018, 484, 298-303, 10.1016/j.cca.2018.06.014.
- Wei Zhang; Jian Song; Yue Zhang; Yingxue Ma; Jing Yang; Guanghui He; Song Chen; Intermittent high glucose-induced oxidative stress modulates retinal pigmented epithelial cell autophagy and promotes cell survival via increased HMGB1. BMC Ophthalmology 2018, 18, 192, 10.1186/s12886-018-0864-5.
- Younan Ren; Shanjun Tao; Shuguo Zheng; Mengqiu Zhao; Yuanmei Zhu; Jieren Yang; Yuanjie Wu; Salvianolic acid B improves vascular endothelial function in diabetic rats with blood glucose fluctuations via suppression of endothelial cell apoptosis. European Journal of Pharmacology 2016, 791, 308-315, 10.1016/j.ejphar.2016.09.014.
- Na Wu; Haitao Shen; Yanjun Wang; Bing He; Yongyan Zhang; Yu Bai; Runyu Du; Qiang Du; Ping Han; Role of the PKCβII/JNK signaling pathway in acute glucose fluctuation-induced apoptosis of rat vascular endothelial cells. International Journal of Earth Sciences 2017, 54, 727-736, 10.1007/s00592-017-0999-5.
- Changjiang Ying; Shanshan Wang; Yan Lu; Lei Chen; Yizhen Mao; Hongwei Ling; Xingbo Cheng; Xiaoyan Zhou; Glucose fluctuation increased mesangial cell apoptosis related to AKT signal pathway.. Archives of Medical Science 2019, 15, 730-737, 10.5114/aoms.2019.84739.
- Qian Chai; Jiajing Miao; Meili Liu; Ziying Zhang; Ziang Meng; Weihua Wu; Knockdown of SGLT1 prevents the apoptosis of cardiomyocytes induced by glucose fluctuation via relieving oxidative stress and mitochondrial dysfunction. Biochemistry and Cell Biology 2021, 99, 356-363, 10.1139/bcb-2020-0491.
- Hui Wang; Jiuling Deng; Liang Chen; Ke Ding; Yi Wang; Acute glucose fluctuation induces inflammation and neurons apoptosis in hippocampal tissues of diabetic rats.. Journal of Cellular Biochemistry 2019, 1, 1– 9, 10.1002/jcb.29523.
- Ting Yan; Zhihui Zhang; Danqing Li; NGF receptors and PI3K/AKT pathway involved in glucose fluctuation-induced damage to neurons and α-lipoic acid treatment. BMC Neuroscience 2020, 21, 1-11, 10.1186/s12868-020-00588-y.
- Cheng-Fang Hsieh; Ching-Kuan Liu; Ching-Tien Lee; Liang-En Yu; Jiz-Yuh Wang; Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Scientific Reports 2019, 9, 1-16, 10.1038/s41598-018-37215-0.
- Bing Xue; Lin Wang; Zhe Zhang; Rui Wang; Xin-Xin Xia; Ping-Ping Han; Li-Jun Cao; Yong-Hui Liu; Lian-Qing Sun; Puerarin may protect against Schwann cell damage induced by glucose fluctuation. Journal of Natural Medicines 2017, 71, 472-481, 10.1007/s11418-016-1067-0.
- Arwa Alnahdi; Annie John; Haider Raza; Mitigation of Glucolipotoxicity-Induced Apoptosis, Mitochondrial Dysfunction, and Metabolic Stress by N-Acetyl Cysteine in Pancreatic β-Cells. Biomolecules 2020, 10, 239, 10.3390/biom10020239.
- Kaliyaperumal Rani; Nway Y. Aung; Docosahexaenoic Acid Inhibits Vascular Smooth Muscle Cell Proliferation Induced by Glucose Variability. The Open Biochemistry Journal 2017, 11, 56-65, 10.2174/1874091X01711010056.
- Sung Hoon Yu; Jae Myung Yu; Hyung Joon Yoo; Seong Jin Lee; Dong Hyun Kang; Young Jung Cho; Doo Man Kim; Anti-Proliferative Effects of Rutin on OLETF Rat Vascular Smooth Muscle Cells Stimulated by Glucose Variability. Yonsei Medical Journal 2016, 57, 373-381, 10.3349/ymj.2016.57.2.373.
- Zhen Zhang; Jing Li; Xinkui Jiang; Lei Yang; Lei Lei; Dehong Cai; Hua Zhang; Hong Chen; GLP-1 ameliorates the proliferation activity of INS-1 cells inhibited by intermittent high glucose concentrations through the regulation of cyclins. Molecular Medicine Reports 2014, 10, 683-688, 10.3892/mmr.2014.2265.
- Nirav Florian Chhabra; Oana Veronica Amarie; Moya Wu; Anna-Lena Amend; Marina Rubey; Daniel Gradinger; Martin Irmler; Johannes Beckers; Birgit Rathkolb; Eckhard Wolf; et al.Annette FeuchtingerPeter HuypensRaffaele TeperinoJan RozmanGerhard K. H. PrzemeckMartin Hrabě de Angelis PAX6 mutation alters circadian rhythm and β cell function in mice without affecting glucose tolerance. Communications Biology 2020, 3, 1-14, 10.1038/s42003-020-01337-x.
- Qinglei Yin; Qicheng Ni; Yichen Wang; Hongli Zhang; Wenyi Li; Aifang Nie; Shu Wang; Yanyun Gu; Qidi Wang; Guang Ning; et al. Raptor determines β-cell identity and plasticity independent of hyperglycemia in mice. Nature Communications 2020, 11, 2538, 10.1038/s41467-020-15935-0.
- Katalin Kiss; Kornélia Baghy; Sándor Spisák; Szilard Szanyi; Zsolt Tulassay; Attila Zalatnai; J.-Matthias Löhr; Ralf Jesenofsky; Ilona Kovalszky; Gábor Firneisz; et al. Chronic Hyperglycemia Induces Trans-Differentiation of Human Pancreatic Stellate Cells and Enhances the Malignant Molecular Communication with Human Pancreatic Cancer Cells. PLOS ONE 2015, 10, e0128059, 10.1371/journal.pone.0128059.
- Simon E. Regnell; Martin J. Hessner; Shuang Jia; Lina Åkesson; Hans Stenlund; Thomas Moritz; Daria La Torre; Åke Lernmark; Longitudinal analysis of hepatic transcriptome and serum metabolome demonstrates altered lipid metabolism following the onset of hyperglycemia in spontaneously diabetic biobreeding rats. PLOS ONE 2017, 12, e0171372, 10.1371/journal.pone.0171372.
- Marina Barreto Felisbino; Mark Ziemann; Ishant Khurana; Jun Okabe; Keith Al-Hasani; Scott Maxwell; K. N. Harikrishnan; Camila Borges Martins de Oliveira; Maria Luiza S. Mello; Assam El-Osta; et al. Valproic acid influences the expression of genes implicated with hyperglycaemia-induced complement and coagulation pathways. Scientific Reports 2021, 11, 1-12, 10.1038/s41598-021-81794-4.
- Maskomani Silambarasan; Jun Rong Tan; Dwi Setyowati Karolina; Arunmozhiarasi Armugam; Charanjit Kaur; Kandiah Jeyaseelan; MicroRNAs in Hyperglycemia Induced Endothelial Cell Dysfunction. International Journal of Molecular Sciences 2016, 17, 518, 10.3390/ijms17040518.
- Vigdis Aas; Nina P. Hessvik; Marianne Wettergreen; Andreas W. Hvammen; Stefan Hallén; G. Hege Thoresen; Arild C. Rustan; Chronic hyperglycemia reduces substrate oxidation and impairs metabolic switching of human myotubes. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2011, 1812, 94-105, 10.1016/j.bbadis.2010.09.014.
- Karanjit Puri; Nathaniel Lal; Rui Shang; Sanjoy Ghosh; Stephane Flibotte; Roger Dyer; Bahira Hussein; Brian Rodrigues; Diabetes Mellitus Severity and a Switch From Using Lipoprotein Lipase to Adipose-Derived Fatty Acid Results in a Cardiac Metabolic Signature That Embraces Cell Death.. Journal of the American Heart Association 2019, 8, e014022, 10.1161/JAHA.119.014022.
- Kristopher G. Maier; Xuan Han; Benjamin Sadowitz; Karen L. Gentile; Frank Middleton; Vivian Gahtan; Thrombospondin-1: A proatherosclerotic protein augmented by hyperglycemia. Journal of Vascular Surgery 2010, 51, 1238-1247, 10.1016/j.jvs.2009.11.073.
- Tran Thi Hien; Karolina M. Turczyńska; Diana Dahan; Mari Ekman; Mario Grossi; Johan Sjögren; Johan Nilsson; Thomas Braun; Thomas Boettger; Eliana Garcia-Vaz; et al.Karin StenkulaKarl SwärdMaria F. GomezSebastian Albinsson Elevated Glucose Levels Promote Contractile and Cytoskeletal Gene Expression in Vascular Smooth Muscle via Rho/Protein Kinase C and Actin Polymerization. Journal of Biological Chemistry 2016, 291, 3552-3568, 10.1074/jbc.m115.654384.
- Torunn Rønningen; Akshay Shah; Andrew H. Reiner; Philippe Collas; Jan Øivind Moskaug; Epigenetic priming of inflammatory response genes by high glucose in adipose progenitor cells. Biochemical and Biophysical Research Communications 2015, 467, 979-986, 10.1016/j.bbrc.2015.10.030.
- Ana Rubin; Anna C. Salzberg; Yuka Imamura; Anzor Grivitishvilli; Joyce Tombran-Tink; Identification of novel targets of diabetic nephropathy and PEDF peptide treatment using RNA-seq. BMC Genomics 2016, 17, 1-17, 10.1186/s12864-016-3199-8.
- Swojani Shrestha; Sandeep Singhal; Donald A. Sens; Seema Somji; Bethany A. Davis; Rachel Guyer; Spencer Breen; Matthew Kalonick; Scott H. Garrett; Elevated glucose represses lysosomal and mTOR-related genes in renal epithelial cells composed of progenitor CD133+ cells. PLOS ONE 2021, 16, e0248241, 10.1371/journal.pone.0248241.
- Patrick Friedrichs; Andrea Schlotterer; Carsten Sticht; Matthias Kolibabka; Paulus Wohlfart; Axel Dietrich; Thomas Linn; Grietje Molema; Hans-Peter Hammes; Hyperglycaemic memory affects the neurovascular unit of the retina in a diabetic mouse model. Diabetologia 2017, 60, 1354-1358, 10.1007/s00125-017-4254-y.
- Shuxin Fan; Ziqi Yang; Yan Liu; Jiawei Zhong; Shuyao Zhang; Yuhua Xiao; Xu Liu; Wei Yi; Chang He; Youjin Hu; et al.Xialin Liu Extensive Sub-RPE Complement Deposition in a Nonhuman Primate Model of Early-Stage Diabetic Retinopathy. Investigative Opthalmology & Visual Science 2021, 62, 30-30, 10.1167/iovs.62.3.30.
- Kondaiah Moganti; Feng Li; Christina Schmuttermaier; Sarah Riemann; Harald Klüter; Alexei Gratchev; Martin C. Harmsen; Julia Kzhyshkowska; Hyperglycemia induces mixed M1/M2 cytokine profile in primary human monocyte-derived macrophages. Immunobiology 2017, 222, 952-959, 10.1016/j.imbio.2016.07.006.
- Kathrin Thiem; Samuel T. Keating; Mihai G. Netea; Niels P. Riksen; Cees J. Tack; Janna Van Diepen; Rinke Stienstra; Hyperglycemic Memory of Innate Immune Cells Promotes In Vitro Proinflammatory Responses of Human Monocytes and Murine Macrophages. The Journal of Immunology 2021, 206, 807-813, 10.4049/jimmunol.1901348.
- Lei Zhong; Agustina Durso; Debra Toiber; Carlos Sebastian; Ryan E. Henry; Douangsone D. Vadysirisack; Alexander Guimaraes; Brett Marinelli; Jakob Wikstrom; Tomer Nir; et al.Clary B. ClishBhavapriya VaitheesvaranOthon IliopoulosIrwin KurlandYuval DorRalph WeisslederOrian S. ShirihaiLeif W. EllisenJoaquin M. EspinosaRaul Mostoslavsky The Histone Deacetylase Sirt6 Regulates Glucose Homeostasis via Hif1α. Cell 2010, 140, 280-293, 10.1016/j.cell.2009.12.041.
- Martine Emery; Natacha Nanchen; Frédéric Preitner; Mark Ibberson; Raphaël Roduit; Biological Characterization of Gene Response to Insulin-Induced Hypoglycemia in Mouse Retina. PLOS ONE 2016, 11, e0150266, 10.1371/journal.pone.0150266.
- Juhye Lena Kim; Edmund F. La Gamma; Todd Estabrook; Necla Kudrick; Bistra B. Nankova; Whole genome expression profiling associates activation of unfolded protein response with impaired production and release of epinephrine after recurrent hypoglycemia. PLOS ONE 2017, 12, e0172789, 10.1371/journal.pone.0172789.
- S. Zervou; Y.-F. Wang; A. Laiho; A. Gyenesei; L. Kytomaki; R. Hermann; S. Abouna; D. Epstein; S. Pelengaris; M. Khan; et al. Short-term hyperglycaemia causes non-reversible changes in arterial gene expression in a fully ‘switchable’ in vivo mouse model of diabetes. Diabetologia 2010, 53, 2676-2687, 10.1007/s00125-010-1887-5.
- Olga V. Saik; Vadim V. Klimontov; Bioinformatic Reconstruction and Analysis of Gene Networks Related to Glucose Variability in Diabetes and Its Complications. International Journal of Molecular Sciences 2020, 21, 8691, 10.3390/ijms21228691.
- Assam El-Osta; Daniella Brasacchio; Dachun Yao; Alessandro Pocai; Peter L. Jones; Robert G. Roeder; Mark E. Cooper; Michael Brownlee; Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. Journal of Experimental Medicine 2008, 205, 2409-2417, 10.1084/jem.20081188.
- Sarah Costantino; Francesco Paneni; Rodolfo Battista; Lorenzo Castello; Giuliana Capretti; Sergio Chiandotto; Luigi Tanese; Giulio Russo; Dario Pitocco; Gaetano A. Lanza; et al.Massimo VolpeThomas F. LüscherFrancesco Cosentino Impact of Glycemic Variability on Chromatin Remodeling, Oxidative Stress, and Endothelial Dysfunction in Patients With Type 2 Diabetes and With Target HbA 1c Levels. Diabetes 2017, 66, 2472-2482, 10.2337/db17-0294.
- Mi-Kyung Kim; Hyesuk Chung; Changsin Yoon; Eunju Lee; Taenyun Kim; Taekyoon Kim; Minjeong Kwon; Soonhee Lee; Byoungdoo Rhee; Jeonghyun Park; et al. Increase of INS-1 cell apoptosis under glucose fluctuation and the involvement of FOXO–SIRT pathway. Diabetes Research and Clinical Practice 2012, 98, 132-139, 10.1016/j.diabres.2012.04.013.
- Erfan Aref-Eshghi; Saumik Biswas; Charlie Chen; Bekim Sadikovic; Subrata Chakrabarti; Glucose-induced, duration-dependent genome-wide DNA methylation changes in human endothelial cells. American Journal of Physiology-Cell Physiology 2020, 319, C268-C276, 10.1152/ajpcell.00011.2020.
- Lobna Abdelzaher; Takahiro Imaizumi; Tokiko Suzuki; Kengo Tomita; Michinori Takashina; Yuichi Hattori; Astaxanthin alleviates oxidative stress insults-related derangements in human vascular endothelial cells exposed to glucose fluctuations. Life Sciences 2016, 150, 24-31, 10.1016/j.lfs.2016.02.087.
- Xinhua Xiao; Yuanyuan Dong; Jing Zhong; Renxian Cao; Xiang Zhao; Gebo Wen; Jianghua Liu; Adiponectin protects endothelial cells from the damages induced by the intermittent high level of glucose. Endocrine 2011, 40, 386-393, 10.1007/s12020-011-9531-9.
- Merlin C. Thomas; Glycemic Exposure, Glycemic Control, and Metabolic Karma in Diabetic Complications. Advances in Chronic Kidney Disease 2014, 21, 311-317, 10.1053/j.ackd.2014.03.004.
- Yinqiong Huang; Zhenling Liao; Xiahong Lin; Xiaohong Wu; Xiaoyu Chen; Xuefeng Bai; Yong Zhuang; Yingxia Yang; Jinying Zhang; Overexpression of miR-146a Might Regulate Polarization Transitions of BV-2 Cells Induced by High Glucose and Glucose Fluctuations. Frontiers in Endocrinology 2019, 10, 719, 10.3389/fendo.2019.00719.
- Bo Wang; Yang Li; Chao You; miR-129-3p Targeting of MCU Protects Against Glucose Fluctuation-Mediated Neuronal Damage via a Mitochondrial-Dependent Intrinsic Apoptotic Pathway. Diabetes, Metabolic Syndrome and Obesity: Targets and Therapy 2021, ume 14, 153-163, 10.2147/dmso.s285179.
- Lucia La Sala; Monica Cattaneo; Valeria De Nigris; Gemma Pujadas; Roberto Testa; Anna Rita Bonfigli; Stefano Genovese; Antonio Ceriello; Oscillating glucose induces microRNA-185 and impairs an efficient antioxidant response in human endothelial cells.. Cardiovascular Diabetology 2016, 15, 71, 10.1186/s12933-016-0390-9.
- Mark E. Cooper; Assam El-Osta; Epigenetics. Circulation Research 2010, 107, 1403-1413, 10.1161/circresaha.110.223552.
- Ammira-Sarah Al-Shabeeb Akil; Laila F. Jerman; Esraa Yassin; Sujitha S. Padmajeya; Alya Al-Kurbi; Khalid A. Fakhro; Reading between the (Genetic) Lines: How Epigenetics is Unlocking Novel Therapies for Type 1 Diabetes. Cells 2020, 9, 2403, 10.3390/cells9112403.
- Xiaoting Luo; Jinzi Wu; Siqun Jing; Liang-Jun Yan; Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity. Aging and disease 2016, 7, 90-110, 10.14336/ad.2015.0702.
- Sachin Suryavanshi; Yogesh A. Kulkarni; NF-κβ: A Potential Target in the Management of Vascular Complications of Diabetes. Frontiers in Pharmacology 2017, 8, 798, 10.3389/fphar.2017.00798.
- Gang Chen; Yufang Chen; Haifeng Chen; Liantao Li; Jin Yao; Qiqin Jiang; Xu Lin; Junping Wen; Lixiang Lin; The effect of NF-κB pathway on proliferation and apoptosis of human umbilical vein endothelial cells induced by intermittent high glucose. Molecular and Cellular Biochemistry 2010, 347, 127-133, 10.1007/s11010-010-0620-5.
- Zhen-Ye Zhang; Ling-Ling Qian; Ning Wang; Ling-Feng Miao; Xin Ma; Shi-Peng Dang; Ying Wu; Xiao-Yu Liu; Xiao-Yan Li; Qiang Chai; et al.Min PanFu YiTian-You LingRu-Xing Wang Glucose fluctuations promote vascular BK channels dysfunction via PKCα/NF-κB/MuRF1 signaling. Journal of Molecular and Cellular Cardiology 2020, 145, 14-24, 10.1016/j.yjmcc.2020.05.021.
- Simon M. Schultze; Brian A. Hemmings; Markus Niessen; Oliver Tschopp; PI3K/AKT, MAPK and AMPK signalling: protein kinases in glucose homeostasis. Expert Reviews in Molecular Medicine 2012, 14, e1, 10.1017/s1462399411002109.
- Zhen-Ye Zhang; Ling-Feng Miao; Ling-Ling Qian; Ning Wang; Miao-Miao Qi; Yu-Min Zhang; Shi-Peng Dang; Ying Wu; Ru-Xing Wang; Molecular Mechanisms of Glucose Fluctuations on Diabetic Complications. Frontiers in Endocrinology 2019, 10, 640, 10.3389/fendo.2019.00640.
- Wei Zhang; Hui Tu Liu; MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Research 2002, 12, 9-18, 10.1038/sj.cr.7290105.
- Joseph L. Evans; Ira D. Goldfine; Betty A. Maddux; Gerold M. Grodsky; Are Oxidative Stress-Activated Signaling Pathways Mediators of Insulin Resistance and -Cell Dysfunction?. Diabetes 2003, 52, 1-8, 10.2337/diabetes.52.1.1.
- Ramon Yarza; Silvia Vela; Maite Solas; Maria J. Ramirez; c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Frontiers in Pharmacology 2016, 6, 321-321, 10.3389/fphar.2015.00321.
- Rosa Bretón-Romero; Bihua Feng; Monika Holbrook; Melissa G. Farb; Jessica L. Fetterman; Erika A. Linder; Brittany D. Berk; Nobuyuki Masaki; Robert M. Weisbrod; Elica Inagaki; et al.Noyan GokceJose J. FusterKenneth WalshNaomi M. Hamburg Endothelial Dysfunction in Human Diabetes Is Mediated by Wnt5a–JNK Signaling. Arteriosclerosis, Thrombosis, and Vascular Biology 2016, 36, 561-569, 10.1161/atvbaha.115.306578.
- Zhimin Tang; Minxiang Lei; [Involvement of JNK signal transduction pathway in endothelial cell apoptosis induced by intermittent high glucose].. Zhong Nan Da Xue Xue Bao Yi Xue Ban 2010, 35, 616–621, 10.3969/j.issn.1672-7347.2010.06.013.
- Zhen-Ye Zhang; Ning Wang; Ling-Ling Qian; Ling-Feng Miao; Shi-Peng Dang; Ying Wu; Ru-Xing Wang; Glucose Fluctuations Promote Aortic Fibrosis through the ROS/p38 MAPK/Runx2 Signaling Pathway.. Journal of Vascular Research 2019, 57, 24-33, 10.1159/000503608.
- Hui Yao Lan; Arthur C.K. Chung; Transforming Growth Factor-β and Smads. Contributions to Nephrology 2011, 170, 75-82, 10.1159/000324949.
- Kuan-Hsing Chen; Cheng-Chieh Hung; Hsiang-Hao Hsu; Yu-Hong Jing; Chih-Wei Yang; Jan-Kan Chen; Resveratrol ameliorates early diabetic nephropathy associated with suppression of augmented TGF-β/smad and ERK1/2 signaling in streptozotocin-induced diabetic rats. Chemico-Biological Interactions 2011, 190, 45-53, 10.1016/j.cbi.2011.01.033.
- Xiaoyun Cheng; Wenke Gao; Yongyan Dang; Xia Liu; Yujuan Li; Xu Peng; Xiyun Ye; Both ERK/MAPK and TGF-Beta/Smad Signaling Pathways Play a Role in the Kidney Fibrosis of Diabetic Mice Accelerated by Blood Glucose Fluctuation. Journal of Diabetes Research 2013, 2013, 1-8, 10.1155/2013/463740.
- X. Ye; X. Cheng; L. Liu; D. Zhao; Y. Dang; Blood glucose fluctuation affects skin collagen metabolism in the diabetic mouse by inhibiting the mitogen-activated protein kinase and Smad pathways. Clinical and Experimental Dermatology 2013, 38, 530-537, 10.1111/ced.12002.
- Sang-Min Jeon; Regulation and function of AMPK in physiology and diseases. Experimental & Molecular Medicine 2016, 48, e245-e245, 10.1038/emm.2016.81.
- Asier González; Michael N. Hall; Sheng-Cai Lin; D. Grahame Hardie; AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metabolism 2020, 31, 472-492, 10.1016/j.cmet.2020.01.015.
- Peng Jiang; Lejiao Ren; Li Zhi; Zhong Yu; Fengxiang Lv; Fengli Xu; Wei Peng; Xiaoyu Bai; Kunlun Cheng; Li Quan; et al.Xiuqin ZhangXianhua WangYan ZhangDan YangXinli HuRui-Ping Xiao Negative regulation of AMPK signaling by high glucose via E3 ubiquitin ligase MG53. Molecular Cell 2021, 81, 629-637.e5, 10.1016/j.molcel.2020.12.008.
- Hong-Yu Zhao, Min Zhao, Tong-Ning Yi, Jin Zhang; Globular adiponectin protects human umbilical vein endothelial cells against apoptosis through adiponectin receptor 1/adenosine monophosphate-activated protein kinase pathway. Chin Med J (Engl) . 2011, 124(16):, 2540-7..
- Catherine E. Gleason; Danhong Lu; Lee A. Witters; Christopher B. Newgard; Morris J. Birnbaum; The Role of AMPK and mTOR in Nutrient Sensing in Pancreatic β-Cells. Journal of Biological Chemistry 2007, 282, 10341-10351, 10.1074/jbc.m610631200.
- Gabriel Leprivier; Barak Rotblat; How does mTOR sense glucose starvation? AMPK is the usual suspect. Cell Death Discovery 2020, 6, 1-5, 10.1038/s41420-020-0260-9.
- Mengqi Li; Chen-Song Zhang; Jin-Wei Feng; Xiaoyan Wei; Cixiong Zhang; Changchuan Xie; Yaying Wu; Simon A. Hawley; Abdelmadjid Atrih; Douglas J. Lamont; et al.Zhichao WangHai-Long PiaoD. Grahame HardieSheng-Cai Lin Aldolase is a sensor for both low and high glucose, linking to AMPK and mTORC1. Cell Research 2020, 31, 478-481, 10.1038/s41422-020-00456-8.
- Muhammad Ali; Shazia Anwer Bukhari; And Han-Woong Lee; Upstream signalling of mTORC1 and its hyperactivation in type 2 diabetes (T2D). BMB Reports 2017, 50, 601-609, 10.5483/bmbrep.2017.50.12.206.
- Zhao Zhong Chong; Kenneth Maiese; Mammalian target of rapamycin signaling in diabetic cardiovascular disease. Cardiovascular Diabetology 2012, 11, 45-45, 10.1186/1475-2840-11-45.
- Yanli Liu; Yarong Zheng; YeKai Zhou; Yi Liu; Mengxuan Xie; Wenjing Meng; Meixia An; The expression and significance of mTORC1 in diabetic retinopathy. BMC Ophthalmology 2020, 20, 1-7, 10.1186/s12886-020-01553-3.
- Mako Yasuda-Yamahara; Shinji Kume; Hiroshi Maegawa; Roles of mTOR in Diabetic Kidney Disease. Antioxidants 2021, 10, 321, 10.3390/antiox10020321.


