 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ahmed Sayed | + 1003 word(s) | 1003 | 2020-07-10 11:42:24 | | | |
| 2 | Ahmed Sayed | Meta information modification | 1003 | 2020-07-11 04:15:36 | | | | |
| 3 | Rita Xu | -160 word(s) | 843 | 2020-07-13 10:41:32 | | | | |
| 4 | Rita Xu | -16 word(s) | 827 | 2020-10-26 09:59:37 | | |
Video Upload Options
The main protease (Mpro) of the newly emerged severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) was subjected to hyphenated pharmacophoric-based and structural-based virtual screenings using a library of microbial natural products (>24,000 compounds). Subsequent filtering of the resulted hits according to Lipinski’s rules was applied to select only the drug-like molecules. Top-scoring hits were further filtered out depending on their ability to show constant good binding affinities towards the molecular dynamic simulation (MDS)-derived enzyme’s conformers. Final MDS experiments were performed on the ligand-protein complexes to verify their binding modes and calculate their binding free energy. Consequently, a final selection of six compounds of microbial origin was proposed to possess high potential as anti-SARS-CoV-2 drug candidates. Our study provides insight into the role of the Mpro structural flexibility during interactions with the possible inhibitors and sheds light on the structure-based design of anti-coronavirus disease 2019 (COVID-19) therapeutics targeting SARS-CoV-2
1. Abstract
The viral genome is predominated by two open reading frames that are connected by a ribosomal frameshift site and encode the two replicase proteins, pp1a and pp1ab [1]. These polyproteins are cleaved by the viral main protease (Mpro), also called chymotrypsin-like protease, 3CLPro [2][3]. Mpro is considered a highly conserved molecular target across coronaviruses, and hence, it was designated as a potential target for anti-coronavirus drug development [4].
2. Introduction
Recently, the microbial-derived FDA-approved anti-parasitic drug ivermectin (12), a semisynthetic pentacyclic sixteen-membered lactone derived from the soil bacterium Streptomyces avermitilis, proved to be an effective in vitro inhibitor of SARS-CoV-2 replication [5][6]. In this context, several in silico techniques that have recently gained a lot of attention in drug discovery campaigns (e.g., structure and ligand-based virtual screening, docking and molecular dynamics) [7][8]. Hence, we initiated a virtual screening of a big library of microbial natural products (more than 24,000 compounds) aimed at the discovery of potential drug candidates against the SARS-CoV-2 Mpro, taking into account the drug-likeness properties to select only druggable candidates. Simple docking protocols do not take into consideration the flexible nature of proteins; therefore, their success rates in most cases are between 60–70% [9]. To increase the docking performance, top hits retrieved from the primary pharmacophore-based screening were further docked on a series of receptor conformers (i.e., ensemble docking) [10] taken from the molecular dynamic simulation (MDS) to consider the factor of the binding pockets’ flexibility. Finally, further MDS of the protein–ligand complexes were performed to verify our docking experiments and calculate their binding free energy (ΔG). Drug candidates proposed in this study could provide a promising starting point for the in vitro and in vivo testing and further development of potential drug leads against the newly emerged coronavirus disease 2019 (COVID-19). The procedure of the current investigation is depicted in Figure 1.
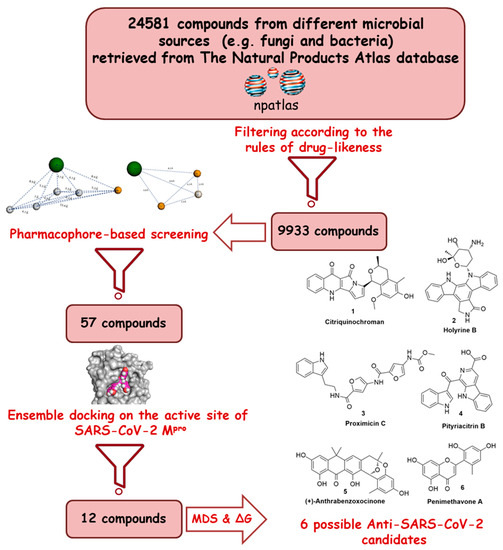
Figure 1. Applied strategy in the present study.
3. Structure and Dynamics of the SARS-CoV-2 Mpro
The catalytic site of the SARS-CoV-2 Mpro was found to be the largest cavity on the whole protein (static volume = 385.56 Å3) and is located in domains I and II (residues 11 to 99 and 100 to 182, respectively, Figure 2A). Additionally, it is smaller than the earlier SARS-CoV Mpro (static volume = 447.7 Å3) [11]. It is worth noting that the enzyme’s domain III, which consists of a globular cluster of five helices, is present only in coronaviruses and is responsible for the regulation of Mpro dimerization [12]. MDS of the reported SARS-CoV-2 Mpro (PDB: 6LU7) [4] was performed to study the conformational changes in the active site using clustering analysis (conformation was sampled every 0.1 ns). The carbon alpha (Cα) root main square deviation (RMSD) values with respect to the initial structure were calculated for 25 ns of the simulation and were found to oscillate from 0.78 to 2.78 Å with a median value of (2.51 Å), reaching equilibrium (i.e., plateau) at 4.8 ns (Figure 2D). Regarding root mean square fluctuation (RMSF) values, they demonstrated that Mpro had moderate flexibility (average RMSF = 2.43 Å, Figure 2E), where the active site showed the highest conformational changes (average RMSF = 3.1 Å), particularly at the THR-45 to ILE-59 loop (Figure 2B), which showed high fluctuations (i.e., RMSF values ranged from 5.2 to 8.9 Å, Figure 2E).
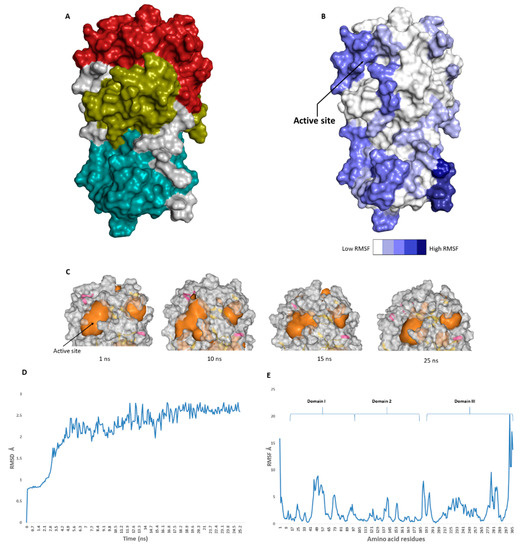
Figure 2. (A): The main domains (Mpro) in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (Brick red: Domain I, Golden yellow: Domain II, Cyan: Domain III), (B): Heat-map illustrating the flexible regions on SARS-CoV-2 Mpro, (C): The active site volume changes during the course of molecular dynamic simulation (MDS) (D and E): root main square deviation (RMSD) and root main square fluctuation (RMSF) of SARS-CoV-2 Mpro after 25 ns of MDS.
The volume of the active site was 399.56 Å3 at the beginning of the simulation (Figure 2C), and during the MDS, it gradually increased to reach 479.65 Å3 at 10 ns, and then began to decrease to reach 314.63 Å3 at the end of the MDS. Docking and virtual screening studies on such flexible catalytic active sites using only their crystalized static form would lead to poor prediction results. Hence, considering multiple structural conformations (i.e., ensemble docking) derived from the MDS study for our virtual screening in the present investigation could significantly improve the predicted results.
References
- Volker Thiel; Konstantin A. Ivanov; Akos Putics; Tobias Hertzig; Barbara Schelle; Sonja Bayer; Benedikt Weißbrich; Eric J. Snijder; Holger Rabenau; Hans Wilhelm Doerr; et al.Alexander E. GorbalenyaJohn Ziebuhr Mechanisms and enzymes involved in SARS coronavirus genome expression. Journal of General Virology 2003, 84, 2305-2315, 10.1099/vir.0.19424-0.
- Ahmed M. Sayed; Amira R. Khattab; Asmaa M. Aboulmagd; Hossam M. Hassan; Mostafa E. Rateb; Hala Zaid; Usama Ramadan Abdelmohsen; Nature as a treasure trove of potential anti-SARS-CoV drug leads: a structural/mechanistic rationale. RSC Advances 2020, 10, 19790-19802, 10.1039/d0ra04199h.
- Markus Hoffmann; Hannah Kleine-Weber; Simon Schroeder; Nadine Krüger; Tanja Herrler; Sandra Erichsen; Tobias S Schiergens; Georg Herrler; Nai-Huei Wu; Andreas Nitsche; et al.Marcel A MüllerChristian DrostenStefan Pöhlmann SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8.
- Zhenming Jin; Xiaoyu Du; Yechun Xu; Yongqiang Deng; Meiqin Liu; Yao Zhao; Bing Zhang; Xiao-Feng Li; Leike Zhang; Chao Peng; et al.Yinkai DuanJing YuLin WangKailin YangFengjiang LiuRendi JiangXinglou YangTian YouXiaoce LiuXiuna YangFang BaiHong LiuXiang LiuLuke W. GuddatWenqing XuGengfu XiaoCheng-Feng QinZheng-Li ShiHualiang JiangZihe RaoHaitao Yang Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020, 582, 289–293, 10.1101/2020.02.26.964882.
- Usama Ramadan Abdelmohsen; Kristina Bayer; Ute Hentschel; Diversity, abundance and natural products of marine sponge-associated actinomycetes. Natural Product Reports 2014, 31, 381-399, 10.1039/c3np70111e.
- Leon Caly; Julian D. Druce; Mike G. Catton; David A. Jans; Kylie M. Wagstaff; The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antiviral Research 2020, 178, 104787, 10.1016/j.antiviral.2020.104787.
- Rahman, N.; Basharat, Z.; Yousuf, M.; Castaldo, G.; Rastrelli, L.; Khan, H; Virtual Screening of Natural Products against Type II Transmembrane Serine Protease (TMPRSS2), the Priming Agent of Coronavirus 2 (SARS-CoV-2). Molecules 2020, 25, 2271.
- Wilfred Ngwa; Rajiv Kumar; Daryl Thompson; William Lyerly; Roscoe Moore; Terry-Elinor Reid; Henry Lowe; Ngeh Toyang; Potential of Flavonoid-Inspired Phytomedicines against COVID-19. Molecules 2020, 25, 2707, 10.3390/molecules25112707.
- Inna Slynko; Michael Schärfe; Tobias Rumpf; Julia Eib; Eric Metzger; Roland Schüle; Manfred Jung; Wolfgang Sippl; Virtual Screening of PRK1 Inhibitors: Ensemble Docking, Rescoring Using Binding Free Energy Calculation and QSAR Model Development. Journal of Chemical Information and Modeling 2014, 54, 138-150, 10.1021/ci400628q.
- Rommie E. Amaro; Jerome Baudry; John Chodera; Özlem Demir; J. Andrew McCammon; Yinglong Miao; Jeremy C. Smith; Ensemble Docking in Drug Discovery. Biophysical Journal 2018, 114, 2271-2278, 10.1016/j.bpj.2018.02.038.
- Davide Gentile; Vincenzo Patamia; Angela Scala; Maria Teresa Sciortino; Anna Piperno; Antonio Rescifina; Putative Inhibitors of SARS-CoV-2 Main Protease from A Library of Marine Natural Products: A Virtual Screening and Molecular Modeling Study. Marine Drugs 2020, 18, 225, 10.3390/md18040225.
- Linlin Zhang; Daizong Lin; Xinyuanyuan Sun; Ute Curth; Christian Drosten; Lucie Sauerhering; Stephan Becker; Katharina Rox; Rolf Hilgenfeld; Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412, 10.1126/science.abb3405.

