 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yoshihisa Matsumoto | + 5616 word(s) | 5616 | 2021-08-04 08:47:28 | | | |
| 2 | Bruce Ren | -21 word(s) | 5595 | 2021-08-12 04:59:32 | | | | |
| 3 | Peter Tang | Meta information modification | 5595 | 2021-10-11 02:51:59 | | |
Video Upload Options
The DNA-dependent protein kinase (DNA-PK) is composed of a DNA-dependent protein kinase catalytic subunit (DNA-PKcs) and Ku70/Ku80 heterodimer. DNA-PK is thought to act as the “sensor” for DNA double-stranded breaks (DSB), which are considered the most deleterious type of DNA damage. In particular, DNA-PKcs and Ku are shown to be essential for DSB repair through nonhomologous end joining (NHEJ). The phenotypes of animals and human individuals with defective DNA-PKcs or Ku functions indicate their essential roles in these developments, especially in neuronal and immune systems. DNA-PKcs are structurally related to Ataxia–telangiectasia mutated (ATM), which is also implicated in the cellular responses to DSBs. DNA-PKcs and ATM constitute the phosphatidylinositol 3-kinase-like kinases (PIKKs) family with several other molecules.
1. DNA-PKcs as a Family Member of ATM
1.1. DNA-PK and DNA-PKcs
1.2. DNA-PKcs and ATM
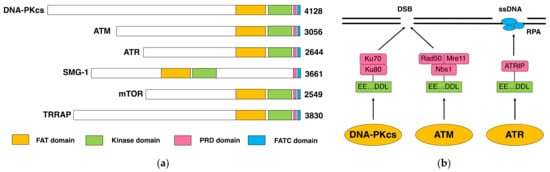
2. Function and Role of DNA-PKcs
2.1. DNA-PKcs in Nonhomologous End Joining
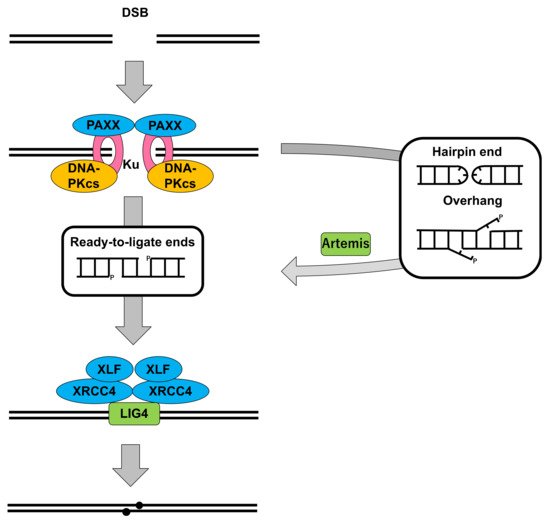
2.2. Cells Deficient for DNA-PKcs: Role in DSB Repair and V(D)J Recombination
| Cell | Species | Type | Mutation 1 | DNA-PKcs Status | Characteristics | Ref. |
|---|---|---|---|---|---|---|
| scid | Mouse | Fibroblast | HMZ c.T12138A, p.Y4046X. (4128 aa) |
Protein very low (~1%); DNA binding undetectable; Kinase activity undetectable. |
Increased IR-sensitivity; Reduced DSB repair ability; Defective V(D)J recombination. | [10][11][12][65][66][67] |
| V3 | Hamster | Ovary | CHTZ; c.C12070A, p.Q4024X; Not known in the other.(4124 aa) |
Protein undetectable; DNA binding barely detectable; Kinase activity undetectable. |
Increased IR-sensitivity; Defective V(D)J recombination. | [10][68] |
| M059J | Human | Glioma | LOH; c.A4051del, p.T1351Pfs*8. (4128 aa) |
Protein undetectable; DNA binding barely detectable; Kinase activity undetectable. |
Increased IR-sensitivity. | [17][69] |
| Irs-20 | Hamster | Ovary | CHTZ c.G12358A, p.E4120K; 2nd allele not expressed.(4124 aa) |
Protein reduced (~10% 2); DNA binding reduced (~25% 2); Kinase activity undetectable. |
Increased IR-sensitivity; Defective V(D)J recombination. | [68][70] |
| SX-9 | Mouse | Mammary carcinoma | CHTZ c.T9572C, p.L3191P; Not known in the other. (4120 aa) |
Protein reduced (~5% 2); DNA binding reduced (~5% 2); Kinase activity undetectable. |
Increased IR-sensitivity; Defective V(D)J recombination. | [70][71] |
| XR-C1 | Hamster | Ovary | Unknown. | Protein undetectable; Kinase activity undetectable. |
Increased IR- and drug (bleomycin and ethyl methane sulfonate) sensitivity; Defective V(D)J recombination. | [72] |
| XR-C2 | Hamster | Ovary | c.G12353A, p.G4118E. (4124 aa) |
Protein expression normal; Kinase activity undetectable. |
Increased IR- and drug (bleomycin, ethyl methane sulfonate and mitomycin C)-sensitivity; Reduced DSB repair ability; Defective V(D)J recombination. | [73][74] |
| (Generated by gene targeting or genome editing) | ||||||
| DT40 | Chicken | B lymphocyte | p. 2888–3012. (4133 aa) |
Protein undetectable. | Normal proliferation; Increased IR-sensitivity. | [75] |
| HCT116 | Human | Colon cancer | p. 3831–4127. (4128 aa) |
Protein undetectable; Kinase activity undetectable. |
Reduced proliferation; Increased IR-, drug (etoposide)-sensitivity; Telomere shortening; Increased chromosomal aberrations. | [76] |
| TK6 | Human | B lymphocyte | Part of exon 32 replaced with drug resistance gene. | Protein undetectable. | Increased IR-sensitivity. | [77] |
| HAP1 | Human | Fibroblast-like, near haploid | 11 bp deletion in exon 25. | Protein undetectable. | Increased drug (Etoposide)-sensitivity. | [78] |
| mESC | Mouse | Embryonic stem | c.24del88; p.R9Wfs7*. (4128 aa) |
mRNA very low (<1%). | Upregulation of pluripotency genes. | [79] |
| HeLa | Human | Cervical carcinoma |
Targeting exon 36. | Protein undetectable. | Increased IR-sensitivity. | [80] |
| Cell | Signal Joint | Coding Joint | Ref. | |||
|---|---|---|---|---|---|---|
| Frequency | Structure | Frequency | Structure | |||
| Mouse scid (SCGR11) | Normal. | Fidelity slightly decreased (~80%). | Significant decrease (~3%). | Larger deletions. | [81] | |
| V3 | Mild decrease (~20%). | Fidelity modestly decreased (~50%). | Significant decrease (~1%). | Abnormally large P elements. | [10] | |
| Mouse scid (SCID/St) | Substantial decrease (~10%). | Not described. | Significant decrease (~0.1%). | Not described. | [11] | |
| IRS-20 | Substantial decrease (~10%). | Fidelity slightly decreased (~75%). | Significant decrease (~3%). | Smaller deletions than scid and V3. | [68] | |
| SX-9 | Substantial decrease (~10%). | Fidelity profoundly decreased (~10%). | Significant decrease (~3%). | Slightly longer deletions. | [71] | |
| XR-C1 | Significant decrease (~2%). | Correct joins absent (0%). | Significant decrease (~2%). | Not described. | [72] | |
| XR-C2 | Mild decrease (~30%). | Not described. | Mild decrease (~50%). | Not described. | [73] | |
| xrs6 ( Ku80) |
Significant decrease (~5%). | Fidelity profoundly decreased (~15%). | Significant decrease (~1%). | None recovered. | [81] | |
| XR-V15B (-Ku80) |
Undetectable (<1%). | Not described. | Undetectable (<1%). | Not described. | [19] | |
| XR-1 (-XRCC4) |
Significant decrease (~2%). | Fidelity profoundly decreased (~20%). | Significant decrease (~0.2%). | Larger deletions. |
|
|
2.3. Animals Deficient for DNA-PKcs: Role in Development
| Animal | Mutation | DNA-PKcs Status | Animal Phenotype | Cellular Phenotype 1 | Ref. |
|---|---|---|---|---|---|
| Mouse scid | c.T12138A, p.Y4046X. (4128 aa) |
Protein very low (~1%); DNA binding undetectable; Kinase activity undetectable. |
SCID; Increased thymic lymphomas. | Increased IR-sensitivity; Reduced DSB repair ability; Defective V(D)J recombination (CJ but not SJ). | [10,11,12,66,67,68] |
| Mouse, gene knockout | Insertion of drug resistance gene in exon 6. | mRNA undetectable; Protein undetectable; Kinase activity undetectable. |
SCID. | Increased IR-sensitivity. | [82] |
| Mouse, gene knockout | p. 3860–3950. (4128 aa) |
Protein undetectable; Kinase activity undetectable. |
SCID. | Defective V(D)J recombination (CJ but not SJ); Increased IR-sensitivity (fibroblast); Normal IR-sensitivity (ES). | [83] |
| Mouse, gene knockout | 3′-half of exon 3 replaced with drug resistance gene. | Protein undetectable. | SCID. | Increased IR-sensitivity. | [84] |
| Mouse slip | A transgene inserted by > 20 copies to upstream of three exons corresponding to 777–1010 nucleotides of mRNA. | mRNA undetectable; Kinase activity undetectable. |
SCID; Increased thymic lymphomas. | Not described. | [85][86] |
| Mouse, KD | c.A11765C, p.D3922A. (4128 aa) |
Protein expression normal; Kinase activity undetectable. |
Embryonic lethal (E14.5); Defective neuronal development. | Increased IR-sensitivity; Increased genomic instability; Defective V(D)J recombination (CJ and SJ). | [87] |
| Mouse, 3A |
c.A7813/7900 /7927G p.T2605/2634/2643A. (4128 aa) |
Protein expression normal; Kinase activity normal. |
Born at normal ratio and size, but becomes smaller 2–3 weeks of age; Death shortly after birth (75% within 4 w); Congenital bone marrow failure; Loss of hematopoietic stem cells. | Increased sensitivity to IR, UV, CPT and MMC. | [88] |
| Mouse, Balb/c, C.B.17, 129 |
c.C6418T, p.R2140C/c.A11530G, p.M3844V. | Protein expression decreased (5–10%); Kinase activity reduced (5–10%). |
Immunologically normal; Normal development; Increased thymocyte apoptosis; Susceptible to cancer, including breast cancer and thymic lymphoma. | Delay in DSB repair; Increased chromosome instability. | [89][90][91] |
| Horse SCID (Arabian foal) |
c.9478del5, p.S3160Nfs4*. (4134 aa) |
Protein undetectable; Kinase activity undetectable. |
SCID. | Increased IR-sensitivity; Defective V(D)J recombination (CJ and SJ). | [92][93][94] |
| Dog SCID (Jack Russel Terriers) |
c.G10879A, p.E3627X. (4144 aa) |
Protein undetectable; DNA binding barely detectable; Kinase activity undetectable. |
SCID. | Increased IR-sensitivity; Defective V(D)J recombination (CJ and SJ). | [95][96] |
| Rat, gene knockout | Deletion in exon 1, causing frame-shift. (4126 aa) |
mRNA undetectable; Protein undetectable. |
SCID; Defective lymphocyte development; Growth retardation; Reduced litter size (~1/2). |
Reduced proliferation; Premature senescence; IR-sensitivity; Defective NHEJ. | [97] |
| Zebrafish, gene knockout | Frame-shift in exon 3. (4119 aa) |
Protein undetectable. | SCID; Growth delay up to 3 months. |
Not described. | [98] |
| Zebrafish, gene knockout | c.10835del8, p.D3612Vfs17. (4119 aa) |
Not described. | SCID; IR-sensitivity. |
Not described. | [99] |
| Mice | Viability | Growth | Neurogenesis | Immunity 1 | Ref. |
|---|---|---|---|---|---|
| DNA-PKcs−/− | Viable. | Normal body size. | Normal. | SCID (leaky); SJ: normal or modestly impaired; CJ: impaired. | [84,85,86,106] |
| Ku80−/− | Viable. | Reduced body size. | Defective (milder than XRCC4−/− and LIG4−/−); Increased cell death. | SCID; SJ & CJ: defective. | [101,102,106] |
| Ku70−/− | Viable. | Reduced body size. | Defective (milder than XRCC4−/− and LIG4−/−); Increased cell death. | SCID (leaky); SJ & CJ: defective. | [103,104,106] |
| LIG4−/− | Late embryonic lethality (>E13.5) | Reduced body size in uterus. | Severely defective; Massive cell death. | SCID; SJ & CJ: defective. | [107][108] |
| XRCC4−/− | Late embryonic lethality (>E13.5) | Reduced body size in uterus. | Severely defective; Massive cell death. | SCID; SJ & CJ: defective. | [109] |
| Artemis−/− | Viable. | Normal body size. | Normal. | SCID (leaky); SJ: normal; CJ: impaired. | [110] |
| XLF−/− | Viable. | Normal body size. | Normal. | Mostly normal; Slight decrease in the number of lymphocytes; Normal lymphocyte distribution; Mild defect in CSR. | [111] |
| PAXX−/− | Viable. | Normal body size. | Normal. | Mostly normal; Modest decrease in the number of lymphocytes. | [112][113] |
2.4. Human Patient and Cells Deficient in DNA-PKcs: Manifested Importance in Human
| Patient | Gender | Ethnic Origin | Mutation 1 and DNA-PK Status | Clinical Characteristics | Cellular Characteristics | Ref. |
|---|---|---|---|---|---|---|
| P1 (ID177) |
F | Turkish | HMZ c.6338del3, p.G2113del/ c.T9185G, p.L3062R. Protein expression normal; Kinase activity normal. |
SCID. | Increased IR-sensitivity; Delay in DSB repair. | [116] |
| P2 (NM720) |
M | British | CHTZ; c. exon16; c.C10721T, p.A3574V. Protein very low (~5% 2); Kinase activity undetectable. |
SCID; Growth failure; Microcephaly; Facial dysmorphism; Seizures; Bilateral sensorineural hearing loss; Visual impairment; Died at 31 months. | Increased IR-sensitivity; Delay in DSB repair. | [119] |
| P3 | M | Turkish | HMZ c.6338del3, p.G2113del/ c.T9185G, p.L3062R. |
SCID; Granuloma; Autoimmunity. | Not described. | [117] |
| P4 | F | Turkish | HMZ c.6338del3, p.G2113del/ c.T9185G, p.L3062R. |
SCID; Granuloma; Autoimmunity. | Not described. | [117] |
| P5 | F | Turkish | HMZ c.6338del3, p.G2113del/ c.T9185G, p.L3062R. |
SCID; Granuloma; Arthritis. | Not described. | [118] |
| P6 | F | Turkish | HMZ c.6338del3, p.G2113del/ c.T9185G, p.L3062R. |
SCID; Granuloma; Diarrhea. | Not described. | [118] |
References
- Walker, A.I.; Hunt, T.; Jackson, R.J.; Anderson, C.W.; Double-stranded DNA induces the phosphorylation of several proteins including the 90 000 mol. wt. heat-shock protein in animal cell extracts.. The EMBO Journal 1985, 4, 139-145, 10.1002/j.1460-2075.1985.tb02328.x.
- Carter, T.; Vancurova, I.; Sun, I.; Lou, W.; DeLeon, S.; A DNA-activated protein kinase from HeLa cell nuclei.. Molecular and Cellular Biology 1990, 10, 6460-6471, 10.1128/mcb.10.12.6460.
- Lees-Miller, S.P.; Chen, Y.R.; Anderson, C.W.; Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Molecular and Cellular Biology 1990, 10, 6472-6481, 10.1128/mcb.10.12.6472-6481.1990.
- Dvir, A.; Stein, L.Y.; Calore, B.L.; Dynan, W.S.; Purification and characterization of a template-associated protein kinase that phosphorylates RNA polymerase II. Journal of Biological Chemistry 1993, 268, 10440-10447, 10.1016/s0021-9258(18)82219-0.
- Gottlieb, T.M.; Jackson, S.P.; The DNA-dependent protein kinase: Requirement for DNA ends and association with Ku antigen. Cell 1993, 72, 131-142, 10.1016/0092-8674(93)90057-w.
- Mimori, T.; Akizuki, M.; Yamagata, H.; Inada, S.; Yoshida, S.; Homma, M.; Characterization of a high molecular weight acidic nuclear protein recognized by autoantibodies in sera from patients with polymyositis-scleroderma overlap.. Journal of Clinical Investigation 1981, 68, 611-620, 10.1172/jci110295.
- Mimori, T.; Hardin, J.A.; Steitz, J.A.; Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders.. Journal of Biological Chemistry 1986, 261, 2274-2278, 10.1016/s0021-9258(17)35929-x.
- Mimori, T.; Hardin, J.A.; Mechanism of interaction between Ku protein and DNA.. Journal of Biological Chemistry 1986, 261, 10375-10379, 10.1016/s0021-9258(18)67534-9.
- Walker, J.R.; Corpina, R.A.; Goldberg, J.; Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature 2001, 412, 607-614, 10.1038/35088000.
- 10. Blunt, T.; Finnie, N.J.; Taccioli, G.E.; Smith, G.C.; Demengeot, J.; Gottlieb, T.M.; Mizuta, R.; Varghese, A.J.; Alt, F.W.; Jeggo, P.A.; et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 1995, 80, 813-823, 10.1016/0092-8674(95)90360-7.
- 11. Kirchgessner, C.U.; Patil, C.K.; Evans, J.W.; Cuomo, C.A.; Fried, L.M.; Carter, T.; Oettinger, M.A.; Brown, J.M.; DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science 1995, 267, 1178-1183, 10.1126/science.7855601.
- Peterson, S.R.; Kurimasa, A.; Oshimura, M.; Dynan, W.S.; Bradbury, E.M.; Chen, D.J.; Loss of the catalytic subunit of the DNA-dependent protein kinase in DNA double-strand-break-repair mutant mammalian cells.. Proceedings of the National Academy of Sciences 1995, 92, 3171-3174, 10.1073/pnas.92.8.3171.
- Bosma, G.C.; Custer, R.P; Bosma, M.J.; A severe combined immunodeficiency mutation in the mouse. Nature 1983, 301, 527-530, 10.1038/301527a0.
- Malynn, B.A.; Blackwell, T.K.; Fulop, G.M.; Rathbun, G.A.; Furley, A.J.W.; Ferrier, P.; Heinke, L.B.; Phillips, R.A; Yancopoulos, G.D.; Alt, F.W.; et al. The scid defect affects the final step of the immunoglobulin VDJ recombinase mechanism. Cell 1988, 54, 453-460, 10.1016/0092-8674(88)90066-9.
- Lieber, M.R.; Hesse, J.E.; Lewis, S.; Bosma, G.C.; Rosenberg, N.; Mizuuchi, K.; Bosma, M.J.; Gellert, M.; The defect in murine severe combined immune deficiency: Joining of signal sequences but not coding segments in V(D)J recombination. Cell 1988, 55, 7-16, 10.1016/0092-8674(88)90004-9.
- Fulop, G.M.; Phillips, R.A.; The scid mutation in mice causes a general defect in DNA repair. Nature 1990, 347, 479-482, 10.1038/347479a0.
- Lees-Miller, S.P.; Godbout, R.; Chan, D.W.; Weinfeld, M.; Day, R.S.3rd.; Barron, G.M.; Allalunis-Turner, J.; Absence of p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science 1995, 267, 1183-1185, 10.1126/science.7855602.
- Taccioli, G.E.; Gottlieb, T.M.; Blunt, T.; Priestley, A.; Demengeot, J.; Mizuta, R.; Lehmann, A.R.; Alt, F.W.; Jackson, S.P.; Jeggo, P.A.; et al. Ku80: product of the XRCC5 gene and its role in DNA repair and V(D)J recombination. Science 1994, 265, 1442-1445, 10.1126/science.8073286.
- Smider, V.; Rathmell, W.K.; Lieber, M.R.; Chu, G.; Restoration of X-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science 1994, 266, 288-291, 10.1126/science.7939667.
- Matsumoto, Y.; Sharma, M.K.; DNA-Dependent protein kinase in DNA damage response: Three decades and beyond. Journal of Radiation and Cancer Research 2020, 11, 123, 10.4103/jrcr.jrcr_60_20.
- Hartley, K.; Gell, D.; Smith, C.; Zhang, H.; Divecha, N.; Connelly, M.; Admon, A.; Lees-Miller, S.; Anderson, C.; Jackson, S.; et al. DNA-dependent protein kinase catalytic subunit: A relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 1995, 82, 849-856, 10.1016/0092-8674(95)90482-4.
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Smith, S.; Uziel, T.; Sfez, S. et al.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749-1753, 10.1126/science.7792600.
- Hari, K.L.; Santerre, A.; Sekelsky, J.J.; McKim, K.S.; Boyd, J.B.; Hawley, R.S.; The mei-41 gene of D. melanogaster is a structural and functional homolog of the human ataxia telangiectasia gene. Cell 1995, 82, 815-821, 10.1016/0092-8674(95)90478-6.
- Greenwell, P.W.; Kronmal, S.L.; Porter, S.E.; Gassenhuber, J.; Obermaier, B.; Petes, T.D.; TEL1, a gene involved in controlling telomere length in S. cerevisiae, is homologous to the human ataxia telangiectasia gene. Cell 1995, 82, 823-829, 10.1016/0092-8674(95)90479-4.
- Morrow, D.M.; Tagle, D.A.; Shiloh, Y.; Collins, F.S.; Hieter, P.; TEL1, an S. cerevisiae homolog of the human gene mutated in ataxia telangiectasia, is functionally related to the yeast checkpoint gene MEC1. Cell 1995, 82, 831-840, 10.1016/0092-8674(95)90480-8.
- Paulovich, A.G.; Hartwell, L.H.; A checkpoint regulates the rate of progression through S phase in S. cerevisiae in Response to DNA damage. Cell 1995, 82, 841-847, 10.1016/0092-8674(95)90481-6.
- Bentley, N.J.; Holtzman, D.A.; Flaggs, G.; Keegan, K.S.; DeMaggio, A.; Ford, J.C.; Hoekstra, M.; Carr, A.M.; The Schizosaccharomyces pombe rad3 checkpoint gene.. The EMBO Journal 1996, 15, 6641-6651.
- Cimprich, K.A.; Shin, T.B.; Keith, C.T.; Schleiber, S.L.; cDNA cloning and gene mapping of a candidate human cell cycle checkpoint protein.. Proceedings of the National Academy of Sciences 1996, 93, 2850-2855, 10.1073/pnas.93.7.2850.
- Denning, G.; Jamieson, L.; Maquat, L.E.; Thompson, E.A.; Fields, A.P.; Cloning of a Novel Phosphatidylinositol Kinase-related Kinase. Journal of Biological Chemistry 2001, 276, 22709-22714, 10.1074/jbc.c100144200.
- Yamashita, A.; Ohnishi, T.; Kashima, I.; Taya, Y.; Ohno, S.; Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes & Development 2001, 15, 2215-2228, 10.1101/gad.913001.
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L.; A mammalian protein targeted by G1-arresting rapamycin–receptor complex. Nature 1994, 369, 756-758, 10.1038/369756a0.
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H.; RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35-43, 10.1016/0092-8674(94)90570-3.
- McMahon, S.B.; Van Buskirk, H.A.; Dugan, K.A.; Copeland, T.D.; Cole, M.D.; The Novel ATM-Related Protein TRRAP Is an Essential Cofactor for the c-Myc and E2F Oncoproteins. Cell 1998, 94, 363-374, 10.1016/s0092-8674(00)81479-8.
- Lees-Miller, S.P.; Anderson, C.W.; The human double-stranded DNA-activated protein kinase phosphorylates the 90-kDa heat-shock protein, hsp90 alpha at two NH2-terminal threonine residues.. Journal of Biological Chemistry 1989, 264, 17275-17280.
- Lees-Miller, S.P.; Sakaguchi, K.; Ullrich, S.J.; Appella, E.; Anderson, C.W.; Human DNA-activated protein kinase phosphorylates serines 15 and 37 in the amino-terminal transactivation domain of human p53. Molecular and Cellular Biology 1992, 12, 5041-5049, 10.1128/mcb.12.11.5041-5049.1992.
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh Y. et al.; et al. Enhanced Phosphorylation of p53 by ATM in Response to DNA Damage. Science 1998, 281, 1674-1677, 10.1126/science.281.5383.1674.
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E., Kastan, M.B.; Siliciano, J.D.; Activation of the ATM Kinase by Ionizing Radiation and Phosphorylation of p53. Science 1998, 281, 1677-1679, 10.1126/science.281.5383.1677.
- Girard, P.M.; Riballo, E.; Begg, A.C.; Waugh, A.; Jeggo P.A.; Nbs1 promotes ATM dependent phosphorylation events including those required for G1/S arrest. Oncogene 2002, 21, 4191-4199, 10.1038/sj.onc.1205596.
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y.; Requirement of the MRN complex for ATM activation by DNA damage. The EMBO Journal 2003, 22, 5612-5621, 10.1093/emboj/cdg541.
- Zou, L.; Elledge, S.J.; Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science 2003, 300, 1542-1548, 10.1126/science.1083430.
- Falck, J.; Coates, J.; Jackson, S.P.; Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605-611, 10.1038/nature03442.
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R.; The molecular basis and disease relevance of non-homologous DNA end joining. Nature Reviews Molecular Cell Biology 2020, 21, 765-781, 10.1038/s41580-020-00297-8.
- Thacker, J.; Zdzienicka, M.Z.; The mammalian XRCC genes: their roles in DNA repair and genetic stability. DNA Repair 2003, 2, 655-672, 10.1016/s1568-7864(03)00062-4.
- Li, Z.; Otevrel, T.; Gao, Y.; Cheng, H.; Seed, B.; Stamato, T.; Taccioli, G.E.; Alt, F.W.; The XRCC4 gene encodes a novel protein involved in DNA double-strand break repair and V(D)J recombination. Cell 1995, 83, 1079-1089, 10.1016/0092-8674(95)90135-3.
- Critchlow, S.; Bowater, R.; Jackson, S.; Mammalian DNA double-strand break repair protein XRCC4 interacts with DNA ligase IV. Current Biology 1997, 7, 588-598, 10.1016/s0960-9822(06)00258-2.
- 47. Grawunder, U.; Wilm, M.; Wu, X.; Kulesza, P.; Wilson, T.; Mann, M.; Lieber, M.R.; Activity of DNA ligase IV stimulated by complex formation with XRCC4 protein in mammalian cells. Nature 1997, 388, 492-495, 10.1038/41358.
- Sado, K.; Ayusawa, D.; Enomoto, A.; Suganuma, T.; Oshimura, M.; Sato, K.; Koyama, H.; Identification of a Mutated DNA Ligase IV Gene in the X-ray-hypersensitive Mutant SX10 of Mouse FM3A Cells. Journal of Biological Chemistry 2001, 276, 9742-9748, 10.1074/jbc.m010530200.
- Riballo, E.; Critchlow, S.E.; Teo, S.H.; Doherty, A.J.; Priestley, A.; Broughton, B.; Kysela, B.; Beamish, H.; Plowman N.; Arlett, C.F.; et al.Lehmann, A.R.Jackson, S.P.Jeggo, P.A. Identification of a defect in DNA ligase IV in a radiosensitive leukaemia patient. Current Biology 1999, 9, 699-S2, 10.1016/s0960-9822(99)80311-x.
- Moshous, D.; Callebaut, I.; de Chasseval, R.; Corneo, B.; Cavazzana-Calvo, M.; Le Deist, F.; Tezcan, I.; Sanal, O.; Bertrand Y.; Philippe, N.; et al.et al. Artemis, a Novel DNA Double-Strand Break Repair/V(D)J Recombination Protein, Is Mutated in Human Severe Combined Immune Deficiency. Cell 2001, 105, 177-186, 10.1016/s0092-8674(01)00309-9.
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R.; Hairpin Opening and Overhang Processing by an Artemis/DNA-Dependent Protein Kinase Complex in Nonhomologous End Joining and V(D)J Recombination. Cell 2002, 108, 781-794, 10.1016/s0092-8674(02)00671-2.
- Buck, D.; Malivert, L.; de Chasseval, R.; Barraud, A.; Fondanèche, M.C.; Sanal, O.; Plebani, A.; Stéphan, J.L.; Hufnagel, M.; le Deist, F. et al.; et al. Cernunnos, a Novel Nonhomologous End-Joining Factor, Is Mutated in Human Immunodeficiency with Microcephaly. Cell 2006, 124, 287-299, 10.1016/j.cell.2005.12.030.
- Ahnesorg, P.; Smith, P.; Jackson, S.P.; XLF Interacts with the XRCC4-DNA Ligase IV Complex to Promote DNA Nonhomologous End-Joining. Cell 2006, 124, 301-313, 10.1016/j.cell.2005.12.031.
- Callebaut, I.; Malivert, L.; Fischer, A.; Mornon, J.P.; Revy, P.; de Villartay, J.P.; Cernunnos Interacts with the XRCC4·DNA-ligase IV Complex and Is Homologous to the Yeast Nonhomologous End-joining Factor Nej1*. Journal of Biological Chemistry 2006, 281, 13857-13860, 10.1074/jbc.c500473200.
- Gu, J.; Lu, H.; Tsai, A.G.; Schwarz, K.; Lieber, M.R.; Single-stranded DNA ligation and XLF-stimulated incompatible DNA end ligation by the XRCC4-DNA ligase IV complex: influence of terminal DNA sequence. Nucleic Acids Research 2007, 35, 5755-5762, 10.1093/nar/gkm579.
- Tsai, C.J.; Kim, S.A.; Chu, G.; Cernunnos/XLF promotes the ligation of mismatched and noncohesive DNA ends. Proceedings of the National Academy of Sciences 2007, 104, 7851-7856, 10.1073/pnas.0702620104.
- Hammel, M.; Yu, Y.; Fang, S.; Lees-Miller, S.P.; Tainer, J.A.; XLF Regulates Filament Architecture of the XRCC4·Ligase IV Complex. Structure 2010, 18, 1431-1442, 10.1016/j.str.2010.09.009.
- Ropars, V.; Drevet, P.; Legrand, P.; Baconnais, S.; Amram, J.; Faure, G.; Márquez, J.A.; Pietrement, O.; Guerois, R.; Callebaut, I. et al.; et al. Structural characterization of filaments formed by human Xrcc4-Cernunnos/XLF complex involved in nonhomologous DNA end-joining. Proceedings of the National Academy of Sciences 2011, 108, 12663-12668, 10.1073/pnas.1100758108.
- Andres, S.N.; Vergnes, A.; Ristic, D.; Wyman, C.; Modesti, M.; Junop, M.; A human XRCC4–XLF complex bridges DNA. Nucleic Acids Research 2012, 40, 1868-1878, 10.1093/nar/gks022.
- Mahaney, B.L.; Hammel, M.; Meek, K.; Tainer, J.A.; Lees-Miller, S.P.; XRCC4 and XLF form long helical protein filaments suitable for DNA end protection and alignment to facilitate DNA double strand break repair. Biochemistry and Cell Biology 2013, 91, 31-41, 10.1139/bcb-2012-0058.
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al.et al. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185-188, 10.1126/science.1261971.
- Xing, M.; Yang, M.; Huo, W.; Feng, F.; Wei, L.; Jiang, W.; Ning, S.; Yan, Z.; Li, W.; Wang, Q.; et al.et al. Interactome analysis identifies a new paralogue of XRCC4 in non-homologous end joining DNA repair pathway. Nature Communications 2015, 6, 6233, 10.1038/ncomms7233.
- Craxton, A.; Somers, J.; Munnur, D.; Jukes-Jones, R.; Cain, K.; Malewicz, M.; XLS (c9orf142) is a new component of mammalian DNA double-stranded break repair. Cell Death & Differentiation 2015, 22, 890-897, 10.1038/cdd.2015.22.
- Kienker, L.J.; Shin, E.K.; Meek, K.; Both V(D)J recombination and radioresistance require DNA-PK kinase activity, though minimal levels suffice for V(D)J recombination. Nucleic Acids Research 2000, 28, 2752-2761, 10.1093/nar/28.14.2752.
- Kurimasa, A.; Kumano, S.; Boubnov, N.; Story, M.; Tung, C.; Peterson, S.; Chen, D.J.; Requirement for the Kinase Activity of Human DNA-Dependent Protein Kinase Catalytic Subunit in DNA Strand Break Rejoining. Molecular and Cellular Biology 1999, 19, 3877-3884, 10.1128/mcb.19.5.3877.
- 66. Blunt, T.; Gell, D.; Fox, M.; Taccioli, G.E.; Lehmann, A.R.; Jackson, S.P.; Jeggo, P.A.; Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the scid mouse.. Proceedings of the National Academy of Sciences 1996, 93, 10285-10290, 10.1073/pnas.93.19.10285.
- Danska, J.S.; Holland, D.P.; Mariathasan, S.; Williams, K.M.; Guidos, C.J.; Biochemical and genetic defects in the DNA-dependent protein kinase in murine scid lymphocytes.. Molecular and Cellular Biology 1996, 16, 5507-5517, 10.1128/mcb.16.10.5507.
- Araki, R.; Fujimori, A.; Hamatani, K.; Mita, K.; Saito, T.; Mori, M.; Fukumura, R.; Morimyo, M.; Muto, M.; Itoh, M. et al.; et al. Nonsense mutation at Tyr-4046 in the DNA-dependent protein kinase catalytic subunit of severe combined immune deficiency mice. Proceedings of the National Academy of Sciences 1997, 94, 2438-2443, 10.1073/pnas.94.6.2438.
- Priestley, A.; Beamish, H.J.; Singleton, B.K.; Blunt, T.; Jeggo, P.A.; Gell, D.; Smith, G.C.M.; Jackson S.P.; Amatucci, A.G et al.; Molecular and biochemical characterisation of DNA-dependent protein kinase-defective rodent mutant irs-20. Nucleic Acids Research 1998, 26, 1965-1973, 10.1093/nar/26.8.1965.
- Anderson, C.W.; Dunn, J.J.; Freimuth, P.I.; Galloway, A.M.; Allalunis-Turner, M.J.; Frameshift Mutation inPRKDC, the Gene for DNA-PKcs, in the DNA Repair-Defective, Human, Glioma-Derived Cell Line M059J. Radiation Research 2001, 156, 2-9, 10.1667/0033-7587(2001)156[0002:fmiptg]2.0.co;2.
- Peterson, S.R; Stackhouse, M.; Waltman, M.J.; Chen, F.; Sato, K.; Chen, D.J.; Characterization of Two DNA Double-stranded Break Repair-deficient Cell Lines That Express Inactive DNA-dependent Protein Kinase Catalytic Subunits. Journal of Biological Chemistry 1997, 272, 10227-10231, 10.1074/jbc.272.15.10227.
- Fukumura, R.; Araki, R.; Fujimori, A.; Mori, M.; Saito, T.; Watanabe, F.; Sarashi, M.; Itsukaichi, H.; Eguchi-Kasai, K.; Sato, K.; et al.et al. Murine Cell Line SX9 Bearing a Mutation in the dna-pkcsGene Exhibits Aberrant V(D)J Recombination Not Only in the Coding Joint but Also in the Signal Joint. Journal of Biological Chemistry 1998, 273, 13058-13064, 10.1074/jbc.273.21.13058.
- Errami, A.; He, D.M.; Friedl, A.A.; Overkamp, W.J.; Morolli, B.; Hendrickson, E.A.; Eckardt-Schupp, F.; Oshimura, M.; Lohman, P.H.; Jackson, S.P. et al.; et al. XR-C1, a new CHO cell mutant which is defective in DNA-PKcs, is impaired in both V(D)J coding and signal joint formation. Nucleic Acids Research 1998, 26, 3146-3153, 10.1093/nar/26.13.3146.
- Errami, A.; Overkamp, W.J.; He, D.M.; Friedl, A.A.; Gell, D.A.; Eckardt-Schupp, F.; Jackson, S.P.; Hendrickson, E.A.; Lohman, P.H.; Zdzienicka, M.Z.; et al. A new X-ray sensitive CHO cell mutant of ionizing radiation group 7, XR-C2, that is defective in DSB repair but has only a mild defect in V(D)J recombination. Mutation Research/DNA Repair 2000, 461, 59-69, 10.1016/s0921-8777(00)00038-0.
- Woods, T.; Wang, W.; Convery, E.; Errami, A.; Zdzienicka, M.Z.; Meek, K.; A single amino acid substitution in DNA-PKcs explains the novel phenotype of the CHO mutant, XR-C2. Nucleic Acids Research 2002, 30, 5120-5128, 10.1093/nar/gkf625.
- Fukushima, T.; Takata, M.; Morrison, C.; Araki, R.; Fujimori, A.; Abe, M.; Tatsumi, K.; Jasin, , M.; Dhar, P. K.; Sonoda, E.; et al.Chi-ba, T.Takeda, S. Genetic Analysis of the DNA-dependent Protein Kinase Reveals an Inhibitory Role of Ku in Late S–G2 Phase DNA Double-strand Break Repair76. Fukushima, T.; Takata, M.; Morrison, C.; Araki, R.; Fujimori, A.; Abe, M.; Tatsumi, K.; Jasin, , M.; Dhar, P. K.; Sonoda, E.; Chi-ba, T.; Takeda, S.. Journal of Biological Chemistry 2001, 276, 44413-44418, 10.1074/jbc.m106295200.
- Ruis, B.L.; Fattah, K.R.; Hendrickson, E.A.; The Catalytic Subunit of DNA-Dependent Protein Kinase Regulates Proliferation, Telomere Length, and Genomic Stability in Human Somatic Cells. Molecular and Cellular Biology 2008, 28, 6182-6195, 10.1128/mcb.00355-08.
- Keka, I.S.; Mohiuddin; Maede, Y.; Rahman, M.M.; Sakuma, T.; Honma, M.; Yamamoto, T.; Takeda, S.; Sasanuma, H.; Smarcal1 promotes double-strand-break repair by nonhomologous end-joining. Nucleic Acids Research 2015, 43, 6359-6372, 10.1093/nar/gkv621.
- Xing, M.; Oksenych, V.; Genetic interaction between DNA repair factors PAXX , XLF, XRCC4 and DNA‐PKcs in human cells. FEBS Open Bio 2019, 9, 1315-1326, 10.1002/2211-5463.12681.
- Soleimani F.; Babaei, E.; Feizi, M.A.H.; Fathi, F.; CRISPR‐Cas9‐mediated knockout of the Prkdc in mouse embryonic stem cells leads to the modulation of the expression of pluripotency genes. Journal of Cellular Physiology 2019, 235, 3994-4000, 10.1002/jcp.29295.
- Douglas, P.; Ye, R.; Radhamani, S.; Cobban, A.; Jenkins, N.P.; Bartlett, E.; Roveredo, J.; Kettenbach, A.N.; Lees-Miller, S.P.; Nocodazole-Induced Expression and Phosphorylation of Anillin and Other Mitotic Proteins Are Decreased in DNA-Dependent Protein Kinase Catalytic Subunit-Deficient Cells and Rescued by Inhibition of the Anaphase-Promoting Complex/Cyclosome with proTAME but Not Apcin. Molecular and Cellular Biology 2020, 40, e00191-19, 10.1128/mcb.00191-19.
- Taccioli, G.E.; Rathbun, G.; Stamato, T.; Jeggo, P.A.; Alt, F.W.; Impairment of V(D)J recombination in double-strand break repair mutants. Science 1993, 260, 207-210, 10.1126/science.8469973.
- Taccioli, G.E.; Amatucci, A.G.; Beamish, H.J.; Gell, D.; Xiang, X.H.; Torres A.M.I; Priestley, A.; Jackson, S.P.; Marshak R.A.; Jeggo P.A.; et al.Herrera, V.L. Targeted Disruption of the Catalytic Subunit of the DNA-PK Gene in Mice Confers Severe Combined Immunodeficiency and Radiosensitivity. Immunity 1998, 9, 355-366, 10.1016/s1074-7613(00)80618-4.
- Gao, Y.; Chaudhuri, J.; Zhu, C.; Davidson, L.; Weaver, D.T.; Alt, F.W.; A Targeted DNA-PKcs-Null Mutation Reveals DNA-PK-Independent Functions for KU in V(D)J Recombination. Immunity 1998, 9, 367-376, 10.1016/s1074-7613(00)80619-6.
- Kurimasa, A.; Ouyang, H.; Dong, L.J.; Wang, S.; Li, X.; Cordon-Cardo C.; Chen, D.J.; Li, G.C.; Catalytic subunit of DNA-dependent protein kinase: Impact on lymphocyte development and tumorigenesis. Proceedings of the National Academy of Sciences 1999, 96, 1403-1408, 10.1073/pnas.96.4.1403.
- Jhappan, C.; Morse H.C.; Fleischmann, R.D.; Gottesman M.M.; Merlino G.; DNA-PKcs: a T-cell tumour suppressor encoded at the mouse scid locus. Nature Genetics 1997, 17, 483-486, 10.1038/ng1297-483.
- Bogue M.A.; Jhappan, C.; Roth D.B.; Analysis of variable (diversity) joining recombination in DNAdependent protein kinase (DNA-PK)-deficient mice reveals DNA-PK-independent pathways for both signal and coding joint formation. Proceedings of the National Academy of Sciences 1998, 95, 15559-15564, 10.1073/pnas.95.26.15559.
- Jiang, W.; Crowe, J.L.; Liu, X.; Nakajima, S.; Wang, Y.; Li, C.; Lee, B.J.; Dubois, R.L.; Liu, C.; Yu, X. et al.; et al. Differential Phosphorylation of DNA-PKcs Regulates the Interplay between End-Processing and End-Ligation during Nonhomologous End-Joining. Molecular Cell 2015, 58, 172-185, 10.1016/j.molcel.2015.02.024.
- Zhang, S.; Yajima, H.; Huynh, H.; Zheng, J.; Callen, N.; Chen H-T.; Wong, N.; Bunting, S.; Lin, Y-F.; Li, M. et al.; et al. Congenital bone marrow failure in DNA-PKcs mutant mice associated with deficiencies in DNA repair. The Journal of Cell Biology 2011, 193, 295-305, 10.1083/jcb.201009074.
- Okayasu R.; Suetomi K.; Yu Y.; Silver A.; Bedford, J.S.; Cox, R.; Ullrich, R.L.; A deficiency in DNA repair and DNA-PKcs expression in the radiosensitive BALB/c mouse.. Cancer Research 2000, 60, 4342-4325.
- Yu, Y.; Okayasu R.; Weil, M.M.; Silver, A.; McCarthy, M.; Zabriskie, R.; Long, S.; Cox, R.; Ullrich, R.L.; Elevated breast cancer risk in irradiated BALB/c mice associates with unique functional polymorphism of the Prkdc (DNA-dependent protein kinase catalytic subunit) gene.. Cancer Research 2001, 61, 1820-1824.
- Mori, N.; Matsumoto, Y.; Okumoto, M.; Suzuki, N; Yamate, J.; Variations in Prkdc encoding the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) and susceptibility to radiation-induced apoptosis and lymphomagenesis. Oncogene 2001, 20, 3609-3619, 10.1038/sj.onc.1204497.
- McGuire, T.C.; Poppie, M.J.; Hypogammaglobulinemia and Thymic Hypoplasia in Horses: a Primary Combined Immunodeficiency Disorder. Infection and Immunity 1973, 8, 272-277, 10.1128/iai.8.2.272-277.1973.
- Wiler, R.; Leber R; Moore, B.B.; VanDyk, L.F.; Perryman, L.E.; Meek, K.; Equine severe combined immunodeficiency: a defect in V(D)J recombination and DNA-dependent protein kinase activity.. Proceedings of the National Academy of Sciences 1995, 92, 11485-11489, 10.1073/pnas.92.25.11485.
- Shin, E.K.; Perryman, L.E.; Meek, K.; A kinase-negative mutation of DNA-PK(CS) in equine SCID results in defective coding and signal joint formation.. The Journal of Immunology 1997, 158, 3565-3569.
- Meek, K; Kienker, L.; Dallas, C.; Wang, W.; Dark, M.J.; Venta, P.J.; Huie, M.L.; Hirschhorn, R.; Bell T.; SCID in Jack Russell Terriers: A New Animal Model of DNA-PKcs Deficiency. The Journal of Immunology 2001, 167, 2142-2150, 10.4049/jimmunol.167.4.2142.
- Ding, Q.; Bramble, L.; Yuzbasiyan-Gurkan, V.; Bell, T.; Meek, K.; DNA-PKcs mutations in dogs and horses: allele frequency and association with neoplasia. Gene 2002, 283, 263-269, 10.1016/s0378-1119(01)00880-0.
- Mashimo, T.; Takizawa, A.; Kobayashi, J.; Kunihiro, Y.; Yoshimi, K.; Ishida, S.; Tanabe, K.; Yanagi, A.; Tachibana, A.; Hirose, J. et al.; et al. Generation and Characterization of Severe Combined Immunodeficiency Rats. Cell Reports 2012, 2, 685-694, 10.1016/j.celrep.2012.08.009.
- Jung, I.H.; Chung, Y.Y.; Jung, D.E.; Kim, Y.J.; Kim do, H.; Kim, K.S.; Park, S.W.; Impaired Lymphocytes Development and Xenotransplantation of Gastrointestinal Tumor Cells in Prkdc -Null SCID Zebrafish Model. Neoplasia 2016, 18, 468-479, 10.1016/j.neo.2016.06.007.
- Moore, J.C.; Tang, Q.; Yordán, N.T.; Moore, F.E.; Garcia E.G; Lobbardi, R.; Ramakrishnan, A.; Marvin, D.L.; Anselmo, A.; Sadreyev, R.I.; et al.Langenau, D.M. Single-cell imaging of normal and malignant cell engraftment into optically clear prkdc-null SCID zebrafish. Journal of Experimental Medicine 2016, 213, 2575-2589, 10.1084/jem.20160378.
- Bosma G.C.; Fried, M.; Custer, R.P.; Carroll, A.; Gibson, D.M.; Bosma, M.J.; Evidence of functional lymphocytes in some (leaky) scid mice.. Journal of Experimental Medicine 1988, 167, 1016-1033, 10.1084/jem.167.3.1016.
- Zhu, C.; Bogue, M.A.; Lim, D.S.; Hasty, P.; Roth, D.B.; Ku86-Deficient Mice Exhibit Severe Combined Immunodeficiency and Defective Processing of V(D)J Recombination Intermediates. Cell 1996, 86, 379-389, 10.1016/s0092-8674(00)80111-7.
- Nussenzweig, A.; Chen, C.; da Costa Soares, V.; Sanchez, M.; Sokol, K.; Nussenzweig, M.C.; Li, G.C.; Requirement for Ku80 in growth and immunoglobulin V(D)J recombination. Nature 1996, 382, 551-555, 10.1038/382551a0.
- Ouyang, H.; Nussenzweig, A.; Kurimasa, A.; Soares, V.C.; Li, X.; Cordon-Cardo C.; Li, W.H.; Cheong, N.; Nussenzweig, M.; Iliakis, G.; et al.Chen, D.J.Li, G.C. Ku70 Is Required for DNA Repair but Not for T Cell Antigen Receptor Gene Recombination In Vivo. Journal of Experimental Medicine 1997, 186, 921-929, 10.1084/jem.186.6.921.
- Gu, Y.; Seidl, K.J.; Rathbun, G.A.; Zhu, C.; Manis, J.P.; van der Stoep, N.; Davidson, L.; Cheng, H.L.; Sekiguchi, J.M; Frank, K.; et al.Stanhope-Baker, P.Schlissel, M.S.Roth, D.BAlt, F.W. Growth Retardation and Leaky SCID Phenotype of Ku70-Deficient Mice. Immunity 1997, 7, 653-665, 10.1016/s1074-7613(00)80386-6.
- Li, G.C.; Ouyang, H.; Li X, Nagasawa H, Little JB, Chen DJ, Ling CC, Fuks Z, Cordon-Cardo C.; Ku70: A Candidate Tumor Suppressor Gene for Murine T Cell Lymphoma. Molecular Cell 1998, 2, 1-8, 10.1016/s1097-2765(00)80108-2.
- Gu, Y.; Sekiguchi, J.; Gao, Y.; Dikkes, P.; Frank, K.; Ferguson, D.; Hasty, P.; Chun, J.; Alt, FW.; Defective embryonic neurogenesis in Ku-deficient but not DNA-dependent protein kinase catalytic subunit-deficient mice. Proceedings of the National Academy of Sciences 2000, 97, 2668-2673, 10.1073/pnas.97.6.2668.
- Frank, K.M.; Sekiguchi, J.M.; Seidl, K.J.; Swat, W.; Rathbun, G.A.; Cheng, H.L.; Davidson, L.; Kangaloo, L.; Alt F.W.; Late embryonic lethality and impaired V (D)J recombination in mice lacking DNA ligase IV. Nature 1998, 396, 173-177, 10.1038/24172.
- Barnes, D.E.; Stamp, G.; Rosewell, I.; Denzel, A.; Lindahl, T.; Targeted disruption of the gene encoding DNA ligase IV leads to lethality in embryonic mice. Current Biology 1998, 8, 1395-1398, 10.1016/s0960-9822(98)00021-9.
- Gao, Y.; Sun, Y.; Frank, K.M.; Dikkes, P.; Fujiwara, Y.; Seidl, K.J.; Sekiguchi, J.M.; Rathbun, G.A.; Swat, W.; Wang, J. et al.; et al. A Critical Role for DNA End-Joining Proteins in Both Lymphogenesis and Neurogenesis. Cell 1998, 95, 891-902, 10.1016/s0092-8674(00)81714-6.
- Rooney, S.; Sekiguchi, J.; Zhu, C.; Cheng, H.L.; Manis, J.; Whitlow, S.; DeVido, J.; Foy, D.; Chaudhuri, J.; Lombard, D. et al.; et al. Leaky Scid Phenotype Associated with Defective V(D)J Coding End Processing in Artemis-Deficient Mice. Molecular Cell 2002, 10, 1379-1390, 10.1016/s1097-2765(02)00755-4.
- Li, G.; Alt, F.W.; Cheng, H.L.; Brush, J.W.; Goff, P.H.; Murphy, M.M.; Franco, S.; Zhang, Y.; Zha, S.; Lymphocyte-Specific Compensation for XLF/Cernunnos End-Joining Functions in V(D)J Recombination. Molecular Cell 2008, 31, 631-640, 10.1016/j.molcel.2008.07.017.
- Kumar, V.; Alt, F.W.; Frock, R.L.; PAXX and XLF DNA repair factors are functionally redundant in joining DNA breaks in a G1-arrested progenitor B-cell line. Proceedings of the National Academy of Sciences 2016, 113, 10619-10624, 10.1073/pnas.1611882113.
- Balmus, G.; Barros, A.C.; Wijnhoven, P.W.; Lescale, C.; Hasse, H.L.; Boroviak, K.; le Sage, C.; Doe, B.; Speak, A.O.; Galli, A. et al.; et al. Synthetic lethality between PAXX and XLF in mammalian development. Genes & Development 2016, 30, 2152-2157, 10.1101/gad.290510.116.
- Zha, S.; Guo, C.; Boboila. C.; Oksenych. V.; Cheng, H.L.; Zhang, Y.; Wesemann, D.R.; Yuen, G.; Patel, H.; Goff, P.H. et al.; et al. ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature 2010, 469, 250-254, 10.1038/nature09604.
- Oksenych, V.; Kumar, V.; Liu, X.; Guo, C.; Schwer, B.; Zha, S.; Alt, F.W.; Functional redundancy between repair factor XLF and damage response mediator 53BP1 in V(D)J recombination and DNA repair. Proceedings of the National Academy of Sciences 2012, 109, 2455-2460, 10.1073/pnas.1121458109.
- van der Burg, M.; Ijspeert, H.; Verkaik, N.S.; Turul, T.; Wiegant, W.W.; Morotomi-Yano, K.; Mari, P.O.; Tezcan, I.; Chen, D.J.; Zdzienicka, M.Z. et al.; et al. A DNA-PKcs mutation in a radiosensitive T–B– SCID patient inhibits Artemis activation and nonhomologous end-joining. Journal of Clinical Investigation 2008, 119, 91-98, 10.1172/jci37141.
- Mathieu, A.L.; Verronese, E.; Rice, G.I.; Fouyssac, F.; Bertrand, Y.; Picard, C.; Chansel, M.; Walter, J.E.; Notarangelo, L.D.; Butte, M.J. et al.; et al. PRKDC mutations associated with immunodeficiency, granuloma, and autoimmune regulator–dependent autoimmunity. Journal of Allergy and Clinical Immunology 2015, 135, 1578-1588, 10.1016/j.jaci.2015.01.040.
- Esenboga, S.; Akal, C.; Karaatmaca, B.; Erman, B.; Dogan, S.; Orhan, D.; Boztug, K.; Ayvaz, D.; Tezcan, İ.; Two siblings with PRKDC defect who presented with cutaneous granulomas and review of the literature. Clinical Immunology 2018, 197, 1-5, 10.1016/j.clim.2018.08.002.
- Woodbine, L.; Neal, J.A.; Sasi, N.K.; Shimada, M.; Deem, K.; Coleman, H.; Dobyns, W.B.; Ogi, T.; Meek, K.; Davies, E.G. et al.; et al. PRKDC mutations in a SCID patient with profound neurological abnormalities. Journal of Clinical Investigation 2013, 123, 2969-2980, 10.1172/jci67349.
- Li, G.; Nelsen, C.; Hendrickson, E.A.; Ku86 is essential in human somatic cells. Proceedings of the National Academy of Sciences 2002, 99, 832-837, 10.1073/pnas.022649699.
- Fattah, F.J.; Lichter, N.F.; Fattah, K.R.; Oh, S.; Hendrickson, E.A.; Ku70, an essential gene, modulates the frequency of rAAV-mediated gene targeting in human somatic cells. Proceedings of the National Academy of Sciences 2008, 105, 8703-8708, 10.1073/pnas.0712060105.
- Wang, Y.; Ghosh, G.; Hendrickson, E.A.; Ku86 represses lethal telomere deletion events in human somatic cells. Proceedings of the National Academy of Sciences 2009, 106, 12430-12435, 10.1073/pnas.0903362106.
- Fattah, F.; Lee, E.H.; Weisensel, N.; Wang, Y.; Lichter, N.; Hendrickson, E.A.; Ku Regulates the Non-Homologous End Joining Pathway Choice of DNA Double-Strand Break Repair in Human Somatic Cells. PLOS Genetics 2010, 6, e1000855-e1000855, 10.1371/journal.pgen.1000855.
- Myung, K.; Ghosh G.; Fattah F.J.; Li G.; Kim H.; Dutia A.; Pak E.; Smith S.; Hendrickson E.A.; Regulation of Telomere Length and Suppression of Genomic Instability in Human Somatic Cells by Ku86. Molecular and Cellular Biology 2004, 24, 5050-5059, 10.1128/mcb.24.11.5050-5059.2004.
- Smith, J.; Riballo, E.; Kysela, B.; Baldeyron, C.; Manolis, K.; Masson, C.; Lieber M.R.; Papadopoulo D.; Jeggo P.; Impact of DNA ligase IV on the fidelity of end joining in human cells. Nucleic Acids Research 2003, 31, 2157-2167, 10.1093/nar/gkg317.
- Katsube, T.; Mori, M.; Tsuji, H.; Shiomi,T .; Shiomi, N.; Onoda M.; Differences in sensitivity to DNA-damaging Agents between XRCC4- and Artemis-deficient human cells.. Journal of Radiation Research 2011, 52, 415-424, 10.1269/jrr.10168.
- Oh, S.; Wang, Y.; Zimbric, J.; Hendrickson, E.A.; Human LIGIV is synthetically lethal with the loss of Rad54B-dependent recombination and is required for certain chromosome fusion events induced by telomere dysfunction. Nucleic Acids Research 2012, 41, 1734-1749, 10.1093/nar/gks1326.
- So, S.; Adachi, N.; Lieber, M.R.; Koyama, H.; Genetic Interactions between BLM and DNA Ligase IV in Human Cells. Journal of Biological Chemistry 2004, 279, 55433-55442, 10.1074/jbc.m409827200.

