 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Marcella Barbarino | + 2109 word(s) | 2109 | 2021-08-11 06:23:42 | | | |
| 2 | Lindsay Dong | Meta information modification | 2109 | 2021-08-12 05:04:29 | | |
Video Upload Options
The expanding clinical application of CDK4- and CDK6-inhibiting drugs in the managements of breast cancer has raised a great interest in testing these drugs in other neoplasms. The potential of combining these drugs with other therapeutic approaches seems to be an interesting work-ground to explore. Even though a potential integration of CDK4 and CDK6 inhibitors with radiotherapy (RT) has been hypothesized, this kind of approach has not been sufficiently pursued, neither in preclinical nor in clinical studies. Similarly, the most recent discoveries focusing on autophagy, as a possible target pathway able to enhance the antitumor efficacy of CDK4 and CDK6 inhibitors is promising but needs more investigations.
1. Introduction
2. The State of the Art in the Clinical Domain
2.1. Clinical Trials of Cyclin-D1/CDK4 and CDK6 Inhibition in Breast Cancer
| Scheme. | INTERVENTION | PATIENT CHARACTERISTICS | MEDIAN PFS(Months) | Clinical Gain/Approval |
|---|---|---|---|---|
| PALOMA-1 | Palbociclib + letrozole vs. letrozole alone |
Post-menopausal women with untreated ER+/HER2−advanced breast cancer | 20.2 vs. 10.2 |
Significant gain in terms of median PFS (10 Months). FDA approval in 2015 |
| PALOMA-2 | Palbociclib + letrozole vs. letrozole alone |
ESR1+/HER2−advanced breast cancer | 27.6 vs. 14.5 |
Delayed ChT: 40.4 vs. 29.9 months |
| PALOMA-3 | Palbociclib + fulvestrant vs. placebo + fulvestrant |
ESR1+/HER2− metastatic breast cancer after hormone therapy (30% received prior ChT) | 9.5 (11.2) vs. 4.6 |
Significant gain in terms of median PFS |
| MONALEESA-2 | Ribociclib + letrozole vs. letrozole alone |
Postmenopausal women with ESR1+/HER2 advanced breast cancer | Not reached vs. 14.7 |
FDA approval in 2017 |
| MONARCH-3 | Abemaciclib + aromatase inhib. vs. placebo + aromatase inhib. |
Postmenopausal women with ESR1+/HER2 locoregionally recurrent or metastatic breast cancer with no prior systemic therapy | 28.1 vs. 14.7 |
Significantly prolonged PFSFDA approval in 2018 |
A metanalysis demonstrated that three CDK4 and CDK6 inhibitors have similar efficacy when associated with AIs, and are superior to either fulvestrant or AI monotherapy independently of any stratification criteria, thus supporting AI plus CDK4 and CDK6 inhibition as the best approach in ESR1+/HER2− metastatic breast cancer [10][12].
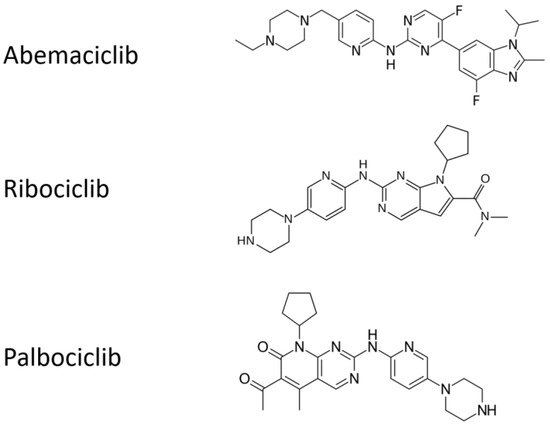
2.2. Real-Life Clinical Reports on Cyclin-D1/CDK4 and CDK6 Inhibitors, Including Association with Radiotherapy
Delivering unplanned RT (mainly with palliative/symptomatic aims) in advanced breast cancer patients on Cyclin-D1/CDK4 and CDK6 inhibitors in prospective trials has produced empirically based treatment not supported by an appropriate rationale and consolidated approach, e g., in terms of RT intent, dose, and scheduling. However, several observational studies provided clearly suggest the lack of additive adverse events when palbociclib or ribociclib are combined with letrozole and concomitant palliative RT [21][22][23]. Additionally, a retrospective single-institution analysis suggested that 15 out a cohort of 42 pts with ESR1+ breast cancer with brain metastases showed promising results using CDK4 and CDK6 inhibitors palbociclib or abemaciclib for 6 months combined with stereotactic radiation [20] and only 5% of the patients developed radiation necrosis. However, only a slight improvement could be shown in survival. However, early radiation toxicities, including esophagitis and severe dermatitis have been reported in a breast cancer patient on palpociclib and palliative RT on cervical lymph nodes [24].
3. Molecular Grounds for Future Developments
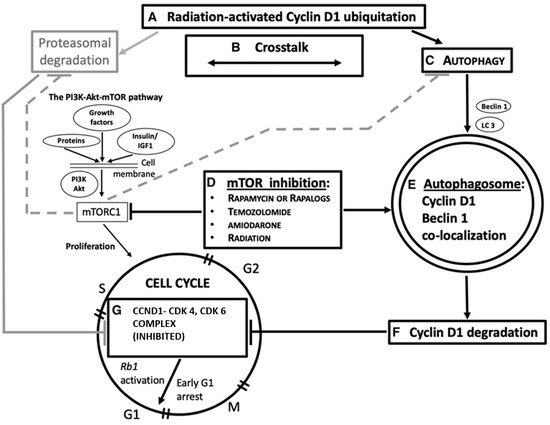
3.1. CCND1 as a Target of Ionizing Radiation in the CCND1-CDK4, and CDK6 Complex
2.2. Ionizing Radiation, Autophagy Enhancement, and CCND1
References
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648.
- Whittaker, S.; Madani, D.; Joshi, S.; Chung, S.A.; Johns, T.; Day, B.; Khasraw, M.; McDonald, K.L. Combination of palbociclib and radiotherapy for glioblastoma. Cell Death Discov. 2017, 3, 17033.
- Nardone, V.; Falivene, S.; Giugliano, F.M.; Gaetano, M.; Giordano, P.; Muto, M.; Daniele, B.; Guida, C. The role of radiation therapy and systemic therapies in elderly with breast cancer. Transl. Cancer Res. 2020, 9, S97–S109.
- Renzulli, M.; Zanotti, S.; Clemente, A.; Mineo, G.; Tovoli, F.; Reginelli, A.; Barile, A.; Cappabianca, S.; Taffurelli, M.; Golfieri, R. Hereditary breast cancer: Screening and risk reducing surgery. Gland. Surg. 2019, 8, S142–S149.
- Sicinska, E.; Aifantis, I.; Le Cam, L.; Swat, W.; Borowski, C.; Yu, Q.; Ferrando, A.A.; Levin, S.D.; Geng, Y.; Von Boehmer, H.; et al. Requirement for cyclin D3 in lymphocyte development and T cell leukemias. Cancer Cell 2003, 4, 451–461.
- Wu, S.Y.; Lan, S.H.; Wu, S.R.; Chiu, Y.C.; Lin, X.Z.; Su, I.J.; Tsai, T.F.; Yen, C.J.; Lu, T.H.; Liang, F.W.; et al. Hepatocellular carcinoma-related cyclin D1 is selectively regulated by autophagy degradation system. Haepatology 2018, 68, 141–154.
- Hamilton, E.; Infante, J.R. Targeting CDK4/6 in patients with cancer. Cancer Treat. Rev. 2016, 45, 129–138.
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666.
- Hortobagyi, G.; Stemmer, S.; Burris, H.; Yap, Y.-S.; Sonke, G.; Paluch-Shimon, S.; Campone, M.; Petrakova, K.; Blackwell, K.; Winer, E.; et al. Updated results from MONALEESA-2, a phase III trial of first-line ribociclib plus letrozole versus placebo plus letrozole in hormone receptor-positive, HER2-negative advanced breast cancer. Ann. Oncol. 2019, 30, 1842.
- Beaver, J.A.; Amiri-Kordestani, L.; Charlab, R.; Chen, W.; Palmby, T.; Tilley, A.; Zirkelbach, J.F.; Yu, J.; Liu, Q.; Zhao, L.; et al. FDA Approval: Palbociclib for the Treatment of Postmenopausal Patients with Estrogen Receptor-Positive, HER2-Negative Metastatic Breast Cancer. Clin. Cancer Res. 2015, 21, 4760–4766.
- Cristofanilli, M.; Turner, N.C.; Bondarenko, I.; Ro, J.; Im, S.-A.; Masuda, N.; Colleoni, M.; DeMichele, A.; Loi, S.; Verma, S.; et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): Final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016, 17, 425–439.
- Turner, N.C.; Ro, J.; André, F.; Loi, S.; Verma, S.; Iwata, H.; Harbeck, N.; Loibl, S.; Huang Bartlett, C.; Zhang, K. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 209–219.
- Asghar, U.; Witkiewicz, A.K.; Turner, N.C.; Knudsen, E.S. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 2015, 14, 130–146.
- Rader, J.; Russell, M.R.; Hart, L.S.; Nakazawa, M.S.; Belcastro, L.T.; Martinez, D.; Li, Y.; Carpenter, E.L.; Attiyeh, E.F.; Diskin, S.J.; et al. Dual CDK4/CDK6 inhibition induces cell-cycle arrest and senescence in neuroblastoma. Clin. Cancer Res. 2013, 19, 6173–6182.
- Slamon, D.J.; Neven, P.; Chia, S.; Fasching, P.A.; De Laurentiis, M.; Im, S.A.; Petrakova, K.; Bianchi, G.V.; Esteva, F.J.; Martín, M.; et al. Overall Survival with Ribociclib plus Fulvestrant in Advanced Breast Cancer. N. Engl. J. Med. 2020, 382, 514–524.
- Tripathy, D.; Im, S.A.; Colleoni, M.; Franke, F.; Bardia, A.; Harbeck, N.; Hurvitz, S.A.; Chow, L.; Sohn, J.; Lee, K.S.; et al. Ribociclib plus endocrine therapy for premenopausal women with hormone-receptor-positive, advanced breast cancer (MONALEESA-7): A randomised phase 3 trial. Lancet Oncol. 2018, 19, 904–915.
- Yang, Y.; Luo, J.; Chen, X.; Yang, Z.; Mei, X.; Ma, J.; Zhang, Z.; Guo, X.; Yu, X. CDK4/6 inhibitors: A novel strategy for tumor radiosensitization. J. Exp. Clin. Cancer Res. 2020, 39, 188.
- Patnaik, A.; Rosen, L.S.; Tolaney, S.M.; Tolcher, A.W.; Goldman, J.W.; Gandhi, L.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Hilton, J.F.; et al. Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov. 2016, 6, 740–753.
- Rossi, V.; Berchialla, P. Should All Patients with HR-Positive HER2-Negative Metastatic Breast Cancer Receive CDK 4/6 Inhibitor as First-Line Based Therapy? A Network Meta-Analysis of Data from the PALOMA 2, MONALEESA 2, MONALEESA 7, MONARCH 3, FALCON, SWOG and FACT trials. Cancers 2019, 11, 1661.
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646.
- Meattini, I.; Desideri, I.; Scotti, V.; Simontacchi, G.; Livi, L. Ribociclib plus letrozole and concomitant palliative radiotherapy for metastatic breast cancer. Breast 2018, 42, 1–2.
- Ratosa, I.; Orazem, M.; Scoccimarro, E.; Steinacher, M.; Dominici, L.; Aquilano, M.; Cerbai, C.; Desideri, I.; Ribnikar, D.; Marinko, T.; et al. Cyclin-Dependent Kinase 4/6 Inhibitors Combined with Radiotherapy for Patients with Metastatic Breast Cancer. Clin. Breast Cancer 2020, 20, 495–502.
- Ippolito, E.; Greco, C.; Silipigni, S.; Dell’Aquila, E.; Petrianni, G.M.; Tonini, G.; Fiore, M.; D’Angelillo, R.M.; Ramella, S. Concurrent radiotherapy with palbociclib or ribociclib for metastatic breast cancer patients: Preliminary assessment of toxicity. Breast 2019, 46, 70–74.
- Li, J.; Huo, X.; Zhao, F.; Ren, D.; Ahmad, R.; Yuan, X.; Du, F.; Zhao, J. Association of Cyclin-Dependent Kinases 4 and 6 Inhibitors with Survival in Patients with Hormone Receptor-Positive Metastatic Breast Cancer: A Systematic Review and Meta-analysis. JAMA Netw. Open 2020, 3, e2020312.
- VanArsdale, T.; Boshoff, C.; Arndt, K.T.; Abraham, R.T. Molecular Pathways: Targeting the Cyclin D-CDK4/6 Axis for Cancer Treatment. Clin. Cancer Res. 2015, 21, 2905–2910.
- Zheng, K.; He, Z.; Kitazato, K.; Wang, Y. Selective Autophagy Regulates Cell Cycle in Cancer Therapy. Theranostics 2019, 9, 104–125.
- Knudsen, E.S.; Witkiewicz, A.K. The Strange Case of CDK4/6 Inhibitors: Mechanisms, Resistance, and Combination Strategies. Trends Cancer 2017, 3, 39–55.
- Casimiro, M.C.; Di Sante, G.; Di Rocco, A.; Loro, E.; Pupo, C.; Pestell, T.G.; Bisetto, S.; Velasco-Velázquez, M.A.; Jiao, X.; Li, Z.; et al. Cyclin D1 Restrains Oncogene-Induced Autophagy by Regulating the AMPK-LKB1 Signaling Axis. Cancer Res. 2017, 77, 3391–3405.
- Shan, J.; Zhao, W.; Gu, W. Suppression of cancer cell growth by promoting cyclin D1 degradation. Mol. Cell 2009, 36, 469–476.
- Choo, D.W.; Baek, H.J.; Motoyama, N.; Cho, K.H.; Kim, H.S.; Kim, S.S. ATM is required for rapid degradation of cyclin D1 in response to gamma-irradiation. Biochem. Biophys. Res. Commun. 2009, 378, 847–850.
- Liu, W.J.; Ye, L.; Huang, W.F.; Guo, L.J.; Xu, Z.G.; Wu, H.L.; Yang, C.; Liu, H.F. p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 2016, 21, 29.
- Paglin, S.; Lee, N.Y.; Nakar, C.; Fitzgerald, M.; Plotkin, J.; Deuel, B.; Hackett, N.; McMahill, M.; Sphicas, E.; Lampen, N.; et al. Rapamycin-sensitive pathway regulates mitochondrial membrane potential, autophagy, and survival in irradiated MCF-7 cells. Cancer Res. 2005, 65, 11061–11070.
- Kim, K.W.; Moretti, L.; Mitchell, L.R.; Jung, D.K.; Lu, B. Endoplasmic reticulum stress mediates radiation-induced autophagy by perk-eIF2alpha in caspase-3/7-deficient cells. Oncogene 2010, 29, 3241–3251.
- De Cataldo, C.; Bruno, F.; Palumbo, P.; Di Sibio, A.; Arrigoni, F.; Clemente, A.; Bafile, A.; Gravina, G.L.; Cappabianca, S.; Barile, A.; et al. Apparent diffusion coefficient magnetic resonance imaging (ADC-MRI) in the axillary breast cancer lymph node metastasis detection: A narrative review. Gland Surg. 2020, 9, 2225–2234.
- Reginelli, A.; Silvestro, G.; Fontanella, G.; Sangiovanni, A.; Conte, M.; Nuzzo, I.; Calvanese, M.; Traettino, M.; Ferraioli, P.; Grassi, R.; et al. Validation of DWI in assessment of radiotreated bone metastases in elderly patients. Int. J. Surg. 2016, 33, S148–S153.
- Mathiassen, S.G.; De Zio, D.; Cecconi, F. Autophagy and the Cell Cycle: A Complex Landscape. Front. Oncol. 2017, 7, 51.
- Brown, N.E.; Jeselsohn, R.; Bihani, T.; Hu, M.G.; Foltopoulou, P.; Kuperwasser, C.; Hinds, P.W. Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res. 2012, 72, 6477–6489.
- Pirtoli, L.; Belmonte, G.; Toscano, M.; Tini, P.; Miracco, C. Cyclin D1 Co-localizes with Beclin-1 in Glioblastoma Recurrences: A Clue to a Therapy-induced, Autophagy-mediated Degradative Mechanism? Anticancer Res. 2016, 36, 4057–4062.
- Chen, G.; Ding, X.F.; Bouamar, H.; Pressley, K.; Sun, L.Z. Everolimus induces G(1) cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells. Am. J. Physiol. Cell. Physiol. 2019, 317, C244–C252.
- Pirtoli, L.; Belmonte, G.; Toscano, M.; Tini, P.; Miracco, C. Comment on “Everolimus induces G1 cell cycle arrest through autophagy-mediated protein degradation of cyclin D1 in breast cancer cells”. Am. J. Physiol. Physiol. 2020, 318, C448–C449.
- Puyol, M.; Martín, A.; Dubus, P.; Mulero, F.; Pizcueta, P.; Khan, G.; Guerra, C.; Santamaría, D.; Barbacid, M. A synthetic lethal interaction between K-Ras oncogenes and Cdk4 unveils a therapeutic strategy for non-small cell lung carcinoma. Cancer Cell. 2010, 18, 63–73.
- Prokhorova, E.A.; Egorshina, A.Y.; Zhivotovsky, B.; Kopeina, G.S. The DNA-damage response and nuclear events as regulators of nonapoptotic forms of cell death. Oncogene 2019, 39, 1–16.
- Pirtoli, L.; Cevenini, G.; Tini, P.; Vannini, M.; Oliveri, G.; Marsili, S.; Mourmouras, V.; Rubino, G.; Miracco, C. The prognostic role of Beclin 1 protein expression in high-grade gliomas. Autophagy 2009, 5, 930–936.
- Matthew-Onabanjo, A.N.; Janusis, J.; Mercado-Matos, J.; Carlisle, A.; Kim, D.; Levine, F.; Cruz-Gordillo, P.; Richards, R.; Lee, M.J.; Shaw, L.M. Beclin 1 Promotes Endosome Recruitment of Hepatocyte Growth Factor Tyrosine Kinase Substrate to Suppress Tumor Proliferation. Cancer Res. 2019, 80, 249–262.

