Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | RAUL SAN MARTIN FACES | + 3023 word(s) | 3023 | 2021-07-26 08:23:55 | | | |
| 2 | Vivi Li | Meta information modification | 3023 | 2021-08-09 04:00:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
San Martin Faces, R. Direct Arylation in Pincer Complexes. Encyclopedia. Available online: https://encyclopedia.pub/entry/12921 (accessed on 22 July 2026).
San Martin Faces R. Direct Arylation in Pincer Complexes. Encyclopedia. Available at: https://encyclopedia.pub/entry/12921. Accessed July 22, 2026.
San Martin Faces, Raul. "Direct Arylation in Pincer Complexes" Encyclopedia, https://encyclopedia.pub/entry/12921 (accessed July 22, 2026).
San Martin Faces, R. (2021, August 08). Direct Arylation in Pincer Complexes. In Encyclopedia. https://encyclopedia.pub/entry/12921
San Martin Faces, Raul. "Direct Arylation in Pincer Complexes." Encyclopedia. Web. 08 August, 2021.
Copy Citation
Direct arylation is an atom-economical alternative to more established procedures such as Stille, Suzuki or Negishi arylation reactions. In comparison with other palladium sources and ligands, the use of palladium pincer complexes as catalysts or pre-catalysts for direct arylation has resulted in improved efficiency, higher reaction yields, and advantageous reaction conditions. In addition to a revision of the literature concerning intra- and intermolecular direct arylation reactions performed in the presence of palladium pincer complexes, the role of these remarkably active catalysts will also be discussed.
direct arylation
palladium
pincer complexes
1. Introduction
Many biologically active compounds contain (hetero)biaryl frameworks. In fact, this pharmacophore core is found in a number of currently prescribed or clinically tested drugs, including several employed in the therapy against cancer or infertility, or antifungal, anti-inflammatory, anti-hypertensive and antibiotic drugs, inter alia (Figure 1) [1][2][3][4][5][6]. In addition to some agrochemical compounds, relevant materials such as liquid crystal displays and molecular switches comprise the (hetero)biaryl motif [7][8][9][10]. Among the methods developed for the preparation of (hetero)biaryl compounds, Ullmann, Scholl, and Gomberg–Bachmann reactions are considered to be classical strategies [11][12][13], whereas palladium or nickel-catalyzed cross-coupling reactions (Suzuki–Miyaura [14], Kumada [15], Stille [16] and Negishi couplings [17]) were discovered at the end of the 20th century and have been extensively utilized due to the large substrate scope and milder conditions involved. Nevertheless, pre-activated or functionalized coupling partners are required for the latter cross-coupling reactions, as (hetero)aryl halides or pseudohalides are coupled with organometallic reagents (organoboron, organomagnesium, organotin, organozinc compounds, respectively). Additional synthetic steps are therefore needed, and the coupling reaction itself often involves the generation of stoichiometric amounts of metal waste. In order to avoid these inconveniences, new methods for (hetero)aryl-aryl bond formation have been devised [18][19][20][21][22][23][24]. In this regard, direct arylation reactions have arisen as a promising alternative to the above cross-coupling strategies. Different names have been applied to define the coupling of a simple (hetero)arene with an aryl halide or pseudohalide. Among them, C–H (bond) activation, cross-dehalogenative coupling, C–H (bond) functionalization, and catalytic direct arylation are generally employed. Although pioneering reports on the use of alternative metals for direct arylation have been published (Cu [25], Fe [26], Ni [27], Ir [28] or Co [29]), second-row transition metals in low oxidation states (Rh [30][31][32][33], Ru [34][35][36][37][38], and especially Pd [39][40][41][42][43]) are preferred as catalysts for these cross-dehalogenative couplings.
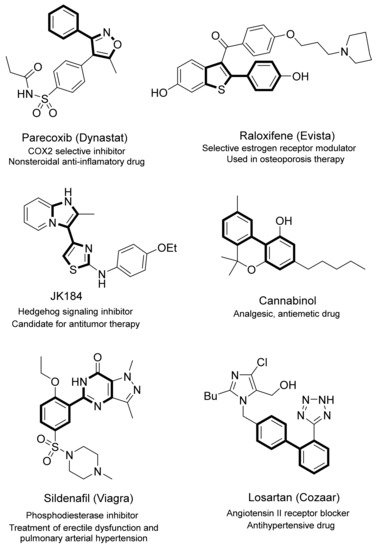
Figure 1. Examples of important bi(hetero)aryl-containing compounds.
The ligands required usually depend on the nature of the haloarene coupling partner. Thus, monodentate triaryl phosphines (e.g., PPh3 and P(o-Tol)3) are typically used for more reactive iodoarenes. Arylation with bromo(hetero)arenes can be also carried out with the same phosphines, although with some substrates better results have been obtained using sterically crowded, electron-rich trialkylphosphanes and biphenylphosphanes [44][45][46][47][48][49][50][51]. The use of chloroarenes in most cross-coupling reactions is often hampered by the more difficult oxidative addition step [52][53]. Therefore, the palladium-catalyzed direct arylation of chloroarenes is usually carried out in the presence of the above trialkyl- and biphenylphosphanes or N-heterocyclic carbenes (NHC) as ligands. Jeffery’s ligand-free conditions have also been successfully used in this field [54][55][56][57][58][59][60][61]. Catalyst loading generally ranges from 1 to 20 mol%.
Alkali carbonates (K2CO3, Cs2CO3), carboxylates (KOAc, CsOPiv) and tBuOK are the bases which are usually employed, although in some cases, bases such as DBU and Et3N have been described. In addition to regenerate the active catalyst, it has been proposed that those bases take part in the formation of diarylpalladium(II) species [62][63][64]. In part due to the higher solubility in organic solvents, Cs2CO3 and CsOPiv have provided better results in some cases. As for solvents, although non-polar toluene and xylene have been employed, N,N-dimethylformamide (DMF), N,N-dimethylacetamide (DMA), acetonitrile, N-methylpyrrolidone (NMP) and dimethylsulfoxide (DMSO) are the commonly used polar aprotic solvents. Heating at temperatures ranging from 100 °C to 140 °C for several hours or days is generally required [65]. Interestingly, a recent report by Albéniz and co-workers demonstrated the beneficial and non-innocent role of alternative solvents such as pinacolone [66].
Several mechanisms have been proposed to explain the direct arylation process. After an initial oxidative addition step, the postulated mechanisms diverge in different pathways. Thus, an electrophilic aromatic substitution-type process might take place [67][68][69], or a concerted termolecular electrophilic substitution [70], or base-assisted intramolecular electrophilic-type substitution [71], or a σ-bond metathesis [72][73], a single electron transfer (SET) [74], or a carbometallation process followed by a β-hydride elimination [75][76], a concerted metalation deprotonation (CMD) [77], or a C–H bond oxidative addition [78][79][80]. Alternatively, in concordance with ruthenium-catalyzed arylations [81][82], a mechanistic pathway based on an initial palladium-catalyzed C–H bond activation has been proposed. As shown in Scheme 1, interaction between the Pd(II) complex and the arene would result in the generation of arylpalladium complex Ar1Pd(II)L, which, upon transmetallation with Ar2X, forms intermediate Ar1Pd(II)Ar2. After reductive elimination of the latter complex with the release of Pd(0) species and Ar1-Ar2, the catalyst would be regenerated by oxidation to Pd(II) [83].
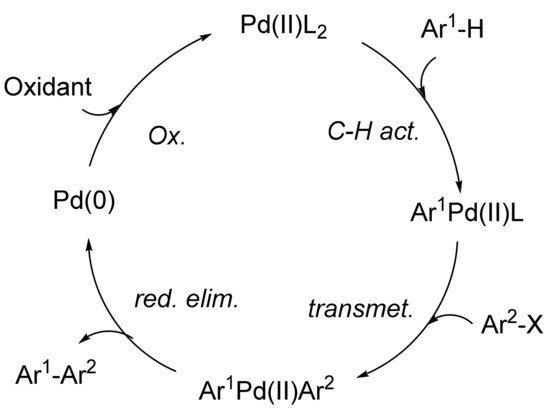
Scheme 1. Alternative pathway to explain palladium-catalyzed direct arylation.
Regioselectivity is often controlled by the electronics of the arene in which C–H functionalization takes place, by the relative C–H acidity, and by the presence of directing groups (nitrogen- or oxygen-coordinating groups, tethering groups, or intramolecular arylations) [84]. As examples of regioselective direct arylation based on the presence of directing groups, Kim and co-workers described the palladium-catalyzed C-8 arylation of dihydroisoquinolones [85]. A regioselective C-3 phenylation of 1-methylquinolin-4(1H)-one was reported by Choi et al. (Scheme 2) [86], and Hartwig’s group presented the regioselective arylation of a number of mono- and disubstituted arenes using synergistic silver and palladium catalysis [87].
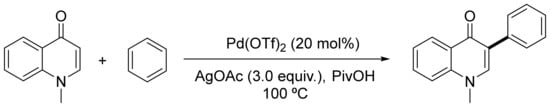
Scheme 2. Regioselective C-3 phenylation of 1-methylquinolin-4(1H)-one.
Direct arylation has been a tool for the construction of several natural products, polycyclic aromatic hydrocarbons (PAHs) and other chemically relevant compounds [88][89][90]. Indeed, the structure of several biologically relevant lactones prepared by direct arylation, including Defucogilocarcins M and E [91][92], and intermediates for the syntheses of Dioncophylline A and Mastigophorene B [93][94][95], are shown in Figure 2. Structurally related Arnottin I was prepared by Harayama and co-workers by direct arylation using Pd(OAc)2 (10 mol%) [96]. The preparation of several korupensamines, i.e., a family of naphthyltetrahydroisoquinoline alkaloids with antimalarial activity, was reported by Bringmann. An intramolecular direct arylation provided the lactone-bridged biaryl displayed in Figure 2, which was atroposelectively cleaved. Arylation employing Herrmann’s catalyst [97] provided a good yield of the required biaryl intermediate, whereas poor results were obtained by using Pd(OAc)2/PPh3. Accordingly, palladacycles can be useful, more efficient palladium sources for direct arylation [98].
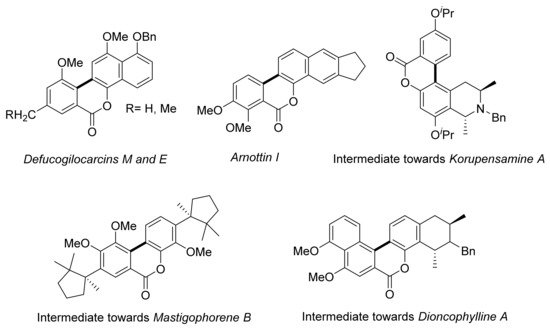
Figure 2. Natural products and synthetic precursors obtained via direct arylation.
Bowl-shaped PAHs are typical synthetic precursors for the access to fullerenes. These fullerene fragments have been prepared in moderate to poor yields by flash vacuum pyrolysis. Moreover, as a result of the harsh reaction conditions needed, only a limited substrate scope is achieved, and this method is difficult to scale up. Following a pioneering report by Rice and co-workers [99], a number of PAHs including bowl-shaped fullerene fragments have been successfully synthesized by the intramolecular direct arylation of o-functionalized biaryl and benzophenanthrene derivatives. High yields and a good tolerance of functional groups were achieved (Scheme 3) [100][101].
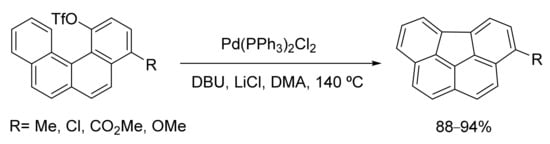
Scheme 3. Intramolecular direct arylation for the synthesis of PAHs.
Moulton and Shaw [102] reported the first examples of pincer complexes in 1976. High thermal, air and moisture stability were exhibited by palladium pincer complexes due to the tight coordination of the tridentate ligand to palladium. Although initially most of these complexes were symmetrical, non-palindromic ligands with hard and soft donor atoms have also been incorporated, thus providing a whole variety of structural designs. Depending on the latter structural features, interaction with substrates and/or the stabilization of reaction intermediates can be facilitated [103][104]. In fact, an increasing number of reports on the application of these terdentate complexes as catalysts or pre-catalysts for a number of synthetic transformations have been published, including recent papers on cross-coupling reactions catalyzed by pincer compounds [105][106][107][108][109][110][111][112][113][114][115][116][117][118][119][120]. In this regard, inter- and intramolecular direct (hetero)arylations have been carried out in the presence of several pincer complexes. This review will cover the literature on the use of palladium pincer complexes for the C–H functionalization of (hetero)arenes with (hetero)aryl halides. A brief summary of the reaction scope, and in some cases, proposals on the role of the pincer complex, will be discussed.
2. Palladium(II) Complexes with Phosphine-Containing Pincer Ligands
Two palladium PCP and PCN complexes were tested as catalysts for the direct access to pyrazolo(benzo)thienoquinolines. This approach involved the intramolecular heteroarylation of 1-aryl-5-(benzo)thienylpyrazoles. The authors confirmed the excellent performance of the above complexes in comparison with commercially available Pd(OAc)2. Indeed, good to excellent yields for the target tetra- and pentacyclic compounds were obtained by using a relatively low amount (1 mol%) of phosphinite- and phosphinoamide-based PCP and PCN complexes. As for Pd(OAc)2, a significantly higher 10 mol% was required to catalyze the same reaction under Jeffrey’s ligand-free conditions, and even then, the yields obtained were lower in all cases. However, no clear differences were found between the catalytic ability of symmetric PCP and non-symmetric PCN complexes (Scheme 4). In addition to this palladium-catalyzed intramolecular heteroarylation, the authors also reported the intermolecular regioselective C-5 arylation of simple 1-substituted thiophenes with an equimolecular amount of bromobenzene under similar conditions [121].
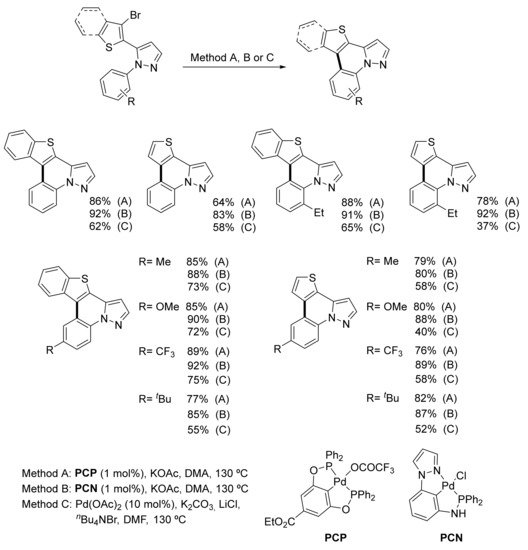
Scheme 4. Synthesis of pyrazolo(benzo)thienoquinolines by direct heteroarylation.
Punji and coworkers reported the intermolecular direct C-2 arylation of benzothiazoles with aryl iodides in the presence of a palladium PCN pincer complex (POCN). 5-Aryloxazoles were also regioselectively arylated at the C-2 position under the same reaction conditions, which involved the use of cesium carbonate as a base and a slight excess of the iodoarene (1.5 equiv.) in DMF at 120 °C. Catalyst loading was optimized at 0.5 mol%, although it was necessary to add CuI (5 mol%) as a co-catalyst. An extensive study on the mechanism of the reaction was carried out using benzothiazole as a model substrate. After observing that the addition of nBu4NBr, a known stabilizer of palladium nanoparticles, did not have a beneficial effect on the reaction outcome, and noticing the results of some poisoning assays and of 31P-NMR monitorization, the authors proposed that, in contrast to previous reports on direct arylation reactions, a Pd(II)–Pd(IV)–Pd(II) pathway could be responsible for the presented arylation. As a result, the authors suggested that the catalytic cycle begins with coordination of benzothiazole with CuX to generate copper complex A, which turns, after H-2 deprotonation, into species B. Alternatively, B could be formed by an initial deprotonation followed by interaction with CuI. Copper-benzothiazolyl complex B would then promote transmetalation with palladium pincer complex PCN (POCN) leading to complex C, which was isolated. Oxidation addition of C with the aryl iodide would generate octahedral Pd(IV) complex D which, upon reductive elimination, would provide the product as well as the initial PCN complex POCN (Scheme 5). Some of the suggested intermediates were isolated and submitted to reaction conditions, providing the expected arylated products. Moreover, these results were corroborated by the kinetic studies and DFT calculations performed by the authors [122][123].
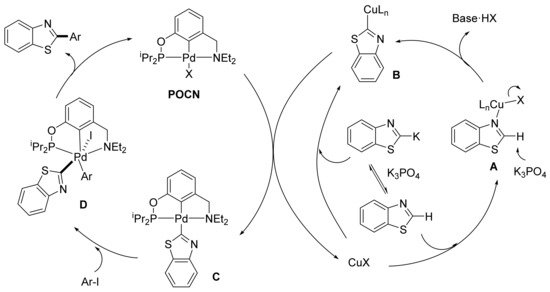
Scheme 5. Mechanistic proposal for the direct heteroarylation of azoles.
As a follow-up research on the results from their previous work on direct heteroarylation [121], in 2015, SanMartin’s group reported the intramolecular direct arylation of amides and sulfonamides in the presence of a PCN palladium pincer complex. The addition of a small amount of water was the key for regioselective access to a wide number of structurally diverse phenanthridinone derivatives using an even smaller amount of catalyst (0.05 mol%) of the phosphinoamide-based PCN palladium pincer depicted in Scheme 6. In addition, benzoisothiazoloindole 5,5-dioxides, benzothiazinoindole 7,7-dioxides, benzopyrroloisothiazole 5,5-dioxides and other biologically relevant sulfur heterocycles were prepared from 2-bromobencenesulfonamides under the same reaction conditions (Scheme 3) [124]. A major advantage of the presented method was the low amount of trace palladium impurities in the final products (0.29 ppm, measured by ICP-MS), certainly due to the small catalyst amount employed. The presence of metal traces in final products is a serious concern for the pharmaceutical industry, with increasingly stricter regulations that limit metal contents to 1 ppm or even less depending on the drug administration route (oral, parenteral, nasal, etc.). In many cases, costly and tedious purification steps are required to remove these toxic contaminants [125]. Wang et al. synthesized a phosphinite-based PCN palladium pincer complex comprising a benzimidazole unit and used it for the direct arylation of (benzo)thiazole and benzoxazole with aryl iodides. As in the paper by Punji [122], CuI was added as a co-catalyst. Using cesium carbonate as a base, they were able to reduce the amount of the palladium catalyst to 0.25 mol%. However, in some cases, 0.5 mol% of the pincer complex and 2.5–5 mol% of CuI were required to attain reasonable yields. A Pd(II)–Pd(IV)–Pd(II) pathway was also proposed to explain the role of the above pincer complex in the arylation reaction [126].
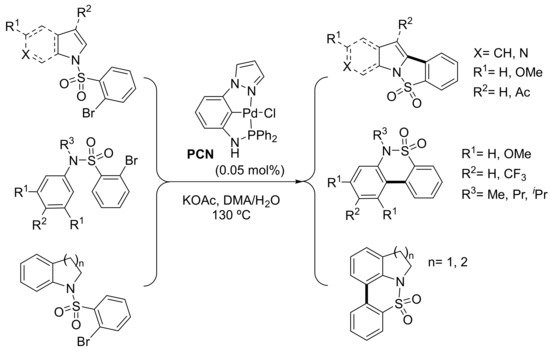
Scheme 6. Intramolecular arylation of o-bromobenzenesulfonamides.
3. NHC Containing Pincer Complexes
N-heterocyclic carbenes (NHC) were introduced in organometallic chemistry by Öfele, who reported the first example back in 1968 [127]. Carbene moieties are usually incorporated in bi- and tridentate ligands due to the high stability they can provide to metal complexes [128][129]. Singh’s group reported the first use of palladium pincer complexes containing NHC moieties as catalysts for direct arylation reactions. After preparing a series of non-palindromic CNS and CNSe complexes (C1–C4) derived from chalcogenated acetamide-functionalized benzimidazolium salts, their catalytic performance in the regioselective C-5 arylation of 1-methyl- and 1,2-dimethylimidazoles with aryl halides under aerobic conditions was examined. A substoichiometric amount of pivalic acid (30 mol%) turned out to be crucial for the reaction outcome. In this regard, the authors proposed that pivalic acid generates coordinatively unsaturated Pd as its proton neutralizes the anionic nitrogen of the amidate fragment, thus helping in the cleavage of the Pd–N bond. That behavior would be consistent with a concerted metalation–deprotonation (CMD) pathway. Benzoic acid was also assayed, providing negligible results. The authors rationalize this outcome considering the higher acidity of benzoic acid, which would stabilize the reaction intermediates so that no further catalysis can occur. As for pincer complexes, 0.5 mol% of catalysts C1–C4 was chosen as optimal for the reaction scope, illustrated by the arylation with sterically hindered 1-bromonaphthalene, as depicted in Scheme 7a. The regioisomeric identity of some of the arylated products was additionally determined by single-crystal X-ray diffraction analysis. Heteroarylation with 3-bromopyridine and 3-bromoquinoline was also carried out under the same conditions. Minor side-products from the C-4 arylation of imidazole and homocoupling of aryl bromides were also detected. 4-Chlorobenzaldehyde and 4-chlorobenzonitrile were also successfully used as arylating agents, although a higher amount of the catalyst (1 mol%) and longer reaction times (20–24 h) were required, probably due to a more difficult oxidative addition step. In addition, the catalytic life of C1–C4 was also tested by recycling or reusing these complexes for six runs. Good yields were obtained in all cases, although a steady decrease was observed in every consecutive run [130]. Very similar reaction conditions (K2CO3, PivOH, DMA, 110 °C) were used by Joshi and co-workers to effect the direct C-5 arylation of imidazole derivatives with aryl bromides in the presence of an SCSe complex (C5), where the NHC moiety occupied the central position of the tridentate ligand (Scheme 7b). Arylation with 4-nitrochlorobenzene was also carried out, although a significant decrease in the reaction yield was observed. In addition, catalyst C5 demonstrated remarkable recyclability up to five consecutive cycles [131]. Following Finke’s report on procedures to distinguish between homogeneous and heterogeneous palladium catalysts [132], both research groups performed poisoning experiments with Hg and PPh3 (Pd/Hg (1/400), 5 mol % of PPh3). Palladium nanoparticles and other palladium (0) species amalgamate with mercury so that a noticeable drop in the conversion yield is observed (mercury drop test). However, no inhibition was observed for the above direct arylation reactions after adding overstoichiometric amounts of these poisoning agents. Considering the results from these poisoning assays and the recyclability exhibited by their pincer complexes, the authors suggested that the catalysis was homogeneous in nature [130][131].
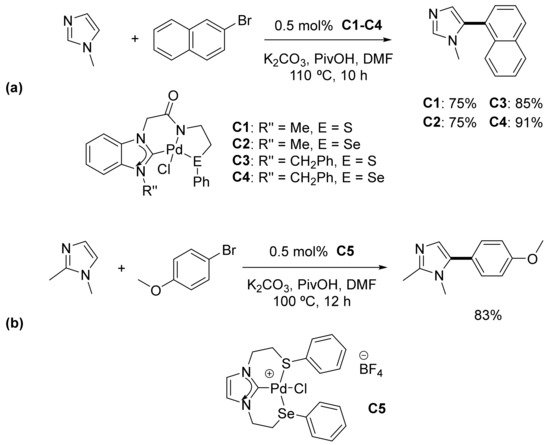
Scheme 7. Regioselective C-5 arylation of 1-methyl- and 1,2-dimethylimidazole derivatives as described by Singh (a) and by Joshi (b).
The selective arylation of 1,2-dimethylimidazole and imidazo[1, 2-a]pyridine derivatives with bromoarenes in the presence of 2 mol% of CNO palladium(II) complexes containing NHC moieties (CNO1–CNO3) was studied by Lee and co-workers (Scheme 8). They also compared their catalytic activity with that of several palladium sources and ligands (PdCl2, Pd(OAc)2, Pd(OAc)2/PCy3, etc.) and found that their CNO complex was less active than their previously reported Pd(0) complex featuring bidentate NHC and PPh2 moieties [133], which could catalyze arylation with chloroarenes. PEPPSI precatalyst Pd(IPr)(3-ClPy)Cl2 [134] also provided the product from the benchmark reaction, the C-5 arylation of 1,2-dimethylimidazole and 4-bromoacetophenone, with equal efficiency. As in many other reports on direct arylation processes, DMA was the solvent of choice, and a significant decrease in the yields was observed when switching to DMF, THF or toluene. Except for 2-bromobenzonitrile and 2-bromotoluene, all the sterically hindered bromoarenes failed to furnish the desired product from 1,2-dimethylimidazole at 140 °C.
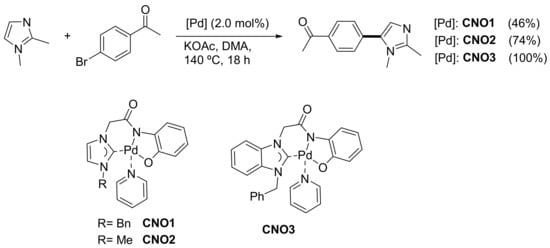
Scheme 8. C-5 arylation of 1,2-dimethylimidazole in the presence of CNO pincer complexes.
In the same paper, imidazo[1,2-a]pyridine was reacted with several bromoarenes bearing electron-donating and electron-withdrawing substituents to provide the corresponding 3-arylated compounds with good to excellent yields. In contrast to 1,2-dimethylimidazole, similar yields were achieved for imidazo[1,2-a]pyridine when the reaction was carried out under argon and under air. 2-Arylbenzothiophenes were also obtained by reactions between thiophene and arylbromides, and in this case, at lower temperature (110 °C, Scheme 9). Gram-scale reactions (10 mmol) between 1,2-dimethylimidazole and 4-bromobenzonitrile, and between imidazo[1.2-a]pyridine and 4-methoxybromobenzene, provided the corresponding arylated compounds in 70% and 66% yields, respectively.
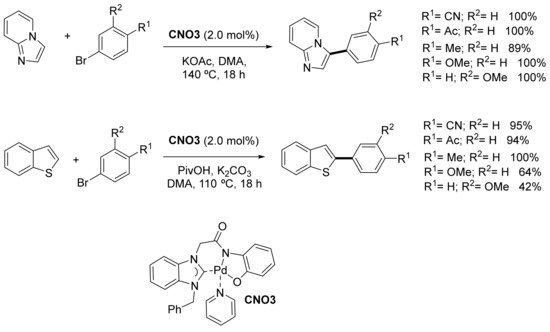
Scheme 9. Direct arylation of imidazo[1,2-a]pyridine and benzothiophene.
The authors also carried out competitive reactions using an equimolecular mixture of 1,2-dimethylimidazole and imidazopyridine and the same bromoarene. After observing that electron-poor imidazopyridine prevailed over electron-rich 1,2-dimethylimidazole (3-arylimidazopyridine was mainly obtained when using 4-bromoanisole, and exclusively isolated when 4-bromoacetophenone was the arylating agent), they suggested that the arylation proceeds via a Pd(II)–Pd(0)–Pd(II) mechanism based on a concerted metalation–deprotonation (CMD) step (Scheme 10). On account of the electron-donating nature of the 1,2-dimethylimidazole unit and the electron-withdrawing character of the imidazo[1.2-a]pyridine core, they synthesized several push–pull chromophores that exhibited a deep blue photoluminescence with moderate quantum efficiency on a large scale, and twisted the intramolecular charge transfer excited state [135].
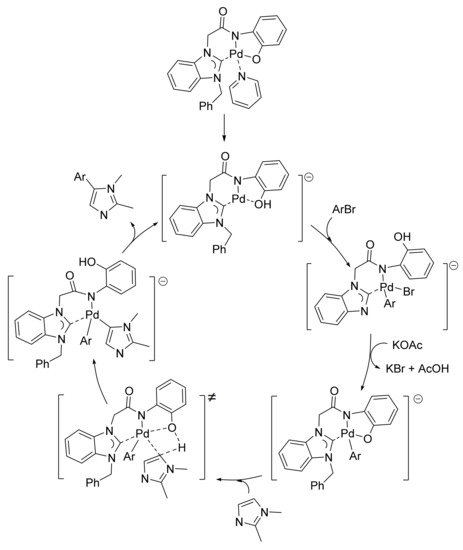
Scheme 10. Mechanism proposed by Lee and co-workers for the C-5 arylation of 1,2-dimethylimidazole.
References
- Polascheck, N.; Bankstahl, M.; Löscher, W. The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp. Neurol. 2010, 224, 219–233.
- Bradley, D.A.; Godfrey, A.G.; Schmid, C.R. Synergistic methodologies for the synthesis of 3-aroyl-2-arylbenzo[b]thiophene-based selective estrogen receptor modulators. Two concise syntheses of raloxifene. Tetrahedron Lett. 1999, 40, 5155–5159.
- Cupido, T.; Rack, P.G.; Firestone, A.J.; Hyman, J.M.; Han, K.; Sinha, S.; Ocasio, C.A.; Chen, J.K. The imidazopyridine derivative JK184 reveals dual roles for microtubules in hedgehog signaling. Angew. Chem. Int. Ed. 2009, 48, 2321–2324.
- Warr, D.; Hesketh, P. Cannabinoids as antiemetics: Everything that’s old is new again. Ann. Oncol. 2020, 31, 1425–1426.
- Dale, D.J.; Dunn, P.J.; Golightly, C.; Hughes, M.L.; Levett, P.C.; Pearce, A.K.; Searle, P.M.; Ward, G.; Wood, A.S. The chemical development of the commercial route to sildenafil: A case history. Org. Proc. Res. Dev. 2000, 4, 17–22.
- Larsen, R.D.; King, A.O.; Chen, C.Y.; Corley, E.G.; Foster, B.S.; Roberts, F.E.; Yang, C.; Lieberman, D.R.; Reamer, R.A.; Tschaen, D.M.; et al. Efficient synthesis of Losartan, a nonpeptide angiotensin II receptor antagonist. J. Org. Chem. 1994, 59, 6391–6394.
- Takale, B.S.; Thakore, R.R.; Mallarapu, R.; Gallou, F.; Lipshutz, B.H. A Sustainable 1-pot, 3-step synthesis of Boscalid using part per Million level Pd catalysis in water. Org. Process Res. Dev. 2020, 24, 101–105.
- Li, Z.; Zhang, X.; Qin, J.; Tan, Z.; Han, M.; Jin, G. Efficient and practical synthesis of 3′,4′,5′-trifluoro-[1,1′-biphenyl]-2-amine: A key intermediate of fluxapyroxad. Org. Process Res. Dev. 2019, 23, 1881–1886.
- Szymborski, T.; Cybulski, O.; Bownik, I.; Żywociński, A.; Wieczorek, S.A.; Fiałkowski, M.; Hołyst, R.; Garstecki, P. Dynamic charge separation in a liquid crystalline meniscus. Soft Matter 2009, 5, 2352–2360.
- Dorazco-Gonzalez, A. Chemosensing of chloride based on a luminescent platinum (II) NCN pincer complex in aqueous media. Organometallics 2014, 33, 868–875.
- Wua, Q.; Wang, L. Immobilization of copper (II) in organic-inorganic hybrid materials: A highly efficient and reusable catalyst for the classic Ullmann reaction. Synthesis 2008, 13, 2007–2012.
- Zani, L.; Dessì, A.; Franchi, D.; Calamante, M.; Reginato, G.; Mordini, A. Transition metal-catalyzed cross-coupling methodologies for the engineering of small molecules with applications in organic electronics and photovoltaics. Coord. Chem. Rev. 2019, 392, 177–236.
- Antenucci, A.; Barbero, M.; Dughera, S.; Ghigo, G. Copper catalysed Gomberg-Bachmann-Hey reactions of arenediazonium tetrafluoroborates and heteroarenediazonium o-benzenedisulfonimides. Synthetic and mechanistic aspects. Tetrahedron 2020, 76, 131632.
- Beletskaya, I.P.; Alonso, F.; Tyurin, V. The Suzuki-Miyaura reaction after the nober prize. Coord. Chem. Rev. 2019, 385, 137–173.
- Iffland, L.; Petuker, A.; van Gastel, M.; Apfel, U.-P. Mechanistic implications for the Ni(I)-catalyzed Kumada cross-coupling reaction. Inorganics 2017, 5, 78.
- Lee, V. Application of copper (I) salt and fluoride promoted Stille coupling reactions in the synthesis of bioactive molecules. Org. Biomol. Chem. 2019, 17, 9095–9123.
- Brittain, W.D.G.; Cobb, S.L. Negishi cross-coupling in the synthesis of amino acids. Org. Biomol. Chem. 2018, 16, 10–20.
- Masui, K.; Ikegami, H.; Mori, A. Palladium-catalyzed C−H homocoupling of thiophenes: facile construction of bithiophene structure. J. Am. Chem. Soc. 2004, 126, 5074–5075.
- Seiple, I.B.; Su, S.; Rodriguez, R.A.; Gianatassio, R.; Fujiwara, Y.; Sobel, A.L.; Baran, P.S. Direct C−H arylation of electron-deficient heterocycles with arylboronic acids. J. Am. Chem. Soc. 2010, 132, 13194–13196.
- Yoshikai, N.; Asako, S.; Yamakawa, T.; Ilies, L.; Nakamura, E. Iron-catalyzed C-H bond activation for the ortho-arylation of aryl pyridines and imines with grignard reagents. Chem. Asian J. 2011, 11, 3059–3065.
- Gorelsky, S.I.; Lapointe, D.; Fagnou, K. Analysis of the palladium-catalyzed (aromatic)C–H bond metalation–deprotonation mechanism spanning the entire spectrum of arenes. J. Org. Chem. 2012, 77, 658–668.
- Sandtorv, A.H. Transition metal-catalyzed C-H activation of indoles. Adv. Synth. Catal. 2015, 357, 2403–2435.
- Castro, L.C.M.; Chatani, N. Nichel catalysts/N,N’-bidentate directing groups: An excellent partnership in directed C-H activation reactions. Chem. Lett. 2015, 44, 410–421.
- Simonetti, M.; Perry, G.J.P.; Cambeiro, X.C.; Juliá-Hernández, F.; Arokianather, J.N.; Larrosa, I. Ru-catalyzed C-H arylation of fluoroarenes with aryl halides. J. Am. Chem. Soc. 2016, 138, 3596–3606.
- Pandey, D.K.; Shabade, A.B.; Punji, B. Copper-catalyzed direct arylation of indoles and related (hetero)arenes: A ligandless and solvent-free approach. Adv. Synth. Catal. 2020, 362, 2534–2540.
- Liu, W.; Cao, H.; Lei, A. Iron-catalyzed direct arylation of unactivated arenes with aryl halides. Angew. Chem. Int. Ed. 2010, 49, 2004–2008.
- Hachiya, H.; Hirano, K.; Satoh, T.; Miura, M. Nichel-catalyzed direct arylation of azoles with aryl bromides. Org. Lett. 2009, 11, 1737–1740.
- Join, B.; Yamamoto, T.; Itami, K. Iridium catalysis for C-H bond arylation of heteroarenes with iodoarenes. Angew. Chem. Int. Ed. 2009, 48, 3644–3647.
- Liu., W.; Cao, H.; Xin, J.; Jin, L.; Lei, A. Cobalt-catalyzed direct arylation of unactivated arenes with aryl halides. Chem. Eur. J. 2011, 17, 3588–3592.
- Berman, A.M.; Lewis, J.C.; Bergman, R.G.; Ellman, J.A. Rh (I)-catalyzed direct arylation of pyridines and quinolones. J. Am. Chem. Soc. 2008, 130, 14926–14927.
- Lewis, J.C.; Berman, A.M.; Bergman, R.G.; Ellman, J.A. Rh (I)-catalyzed arylation of heterocycles via C-H bond activation: Expanded scope through mechanistic insight. J. Am. Chem. Soc. 2008, 130, 2493–2500.
- Berman, A.M.; Bergaman, R.G.; Ellman, J.A. Rh (I)-catalyzed direct arylation of azines. J. Org. Chem. 2010, 75, 7863–7868.
- Wang, Q.; Cai, Z.-J.; Liu, C.-X.; Gu, Q.; You, S.-L. Rhodium-catalyzed atroposelective C-H arylation: Efficient synthesis of axially chiral heterobiaryls. J. Am. Chem. Soc. 2019, 141, 9504–9510.
- Ackermann, L.; Vicente, R.; Potukuchi, H.K.; Pirovano, V. Mechanistic insight into direct arylation with ruthenium (II) carboxylate catalysts. Org. Lett. 2010, 12, 5032–5035.
- Al Mamari, H.H.; Diers, E.; Ackermann, L. Triazole-assisted ruthenium-catalyzed C-H arylarion of aromatic amides. Chem. Eur. J. 2014, 20, 9739–9743.
- Roman, D.S.; Poiret, V.; Pelletier, G.; Charette, A.B. Direct arylation of imidazo[1,5-a]azines through ruthenium and palladium catalysis. Eur. J. Org. Chem. 2015, 2015, 67–71.
- Zha, G.-F.; Qin, H.-L.; Kantchev, E.A.B. Ruthenium-catalyzed direct arylations with aryl chlorides. RSC Adv. 2016, 6, 30875–30885.
- Kaloğlu, N.; Özdemir, İ.; Gürbüz, N.; Arslan, H.; Dixneuf, P.H. Ruthenium (η6, η1-arene-CH2-NHC) catalysts for direct arylation of 2-phenylpyridine with (hetero)aryl chlorides in water. Molecules 2018, 23, 647.
- Lafrance, M.; Rowley, C.N.; Woo, T.K.; Fagnou, K. Catalytic intermolecular direct arylation of perfluorobenzenes. J. Am. Chem. Soc. 2006, 128, 8754–8756.
- Satoh, T.; Miura, M. Catalytic direct arylation of heteroaromatic compounds. Chem. Lett. 2005, 36, 200–205.
- Yuen, O.Y.; Leung, M.P.; Chau Ming So, C.M.; Sun, R.W.-Y.; Kwong, F.Y. Palladium-Catalyzed Direct Arylation of Polyfluoroarenes for Accessing Tetra-ortho-Substituted Biaryls: Buchwald-type Ligand Having Complementary −PPh2 Moiety Exhibits Better Efficiency. J. Org. Chem. 2018, 83, 9008–9017.
- Bheeter, C.B.; Bera, J.K.; Doucet, H. Palladium-catalyzed direct arylation of thiophenes bearing SO2R substituents. J. Org. Chem. 2011, 76, 6407–6413.
- Hayashi, S.; Kojima, Y.; Koizumi, T. Highly regioselective Pd/C-catalyzed direct arylation toward thiphene-based-conjugated polymers. Polym. Chem. 2015, 6, 881–885.
- Yokooji, A.; Okazawa, T.; Satoh, T.; Miura, M.; Nomura, M. Palladium-catalyzed direct arylation of thiazoles with aryl bromides. Tetrahedron Lett. 2003, 59, 5685–5689.
- Turner, G.L.; Morris, J.A.; Greaney, M.F. Direct arylation of thiazoles on water. Angew. Chem. Int. Ed. 2007, 46, 7996–8000.
- Ackermann, L.; Vicente, R.; Born, R. Palladium-catalyzed direct arylations of 1,2,3-triazoles with aryl chlorides using conventional heating. Adv. Synth. Catal. 2008, 350, 741–748.
- Ohnmacht, S.A.; Culshaw, A.J.; Greaney, M.F. Direct arylation of 2H-Indazoles on water. Org. Lett. 2010, 12, 224–226.
- Ben-Yahia, A.; Naas, M.; El Kazzouli, S.; Essassi, M.; Guillaumet, G. Direct C-3-arylations of 1H-Indazoles. Eur. J. Org. Chem. 2012, 2012, 7075–7081.
- Carrër, A.; Brinet, D.; Florent, J.-C.; Rousselle, P.; Bertounesque, E. Palladium-catalyzed direct arylation of polysubstituted benzofurans. J. Org. Chem. 2012, 77, 1316–1327.
- Rampon, D.S.; Wssjohann, L.A.; Schneider, P.H. Palladium-catalyzed direct arylation of selenophene. J. Org. Chem. 2014, 79, 5987–5992.
- Bedford, R.B.; Durrat, S.J.; Montgomery, M. Catalyst-Switchable regiocontrol in the direct arylation of remote C-H groups in pyrazolo[1,5-a]pyrimidines. Angew. Chem. Int. Ed. 2015, 54, 8787–8790.
- Kumar, P.V.; Lin, W.-S.; Shen, J.-S.; Nandi, D.; Lee, H.M. Direct C5-arylation reaction between imidazoles and aryl chlorides catalysed by palladium complexes with phosphines and N-heterocyclic carbenes. Organometallics 2011, 30, 5160–5169.
- Littke, A.F.; Fu, G.C. Palladium-catalyzed couplikng reactions of aryl chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176–4211.
- Campeau, L.-C.; Thansandote, P.; Fagnou, K. High-yielding intramolecular direct arylation reactions with aryl chlorides. Org. Lett. 2005, 7, 1857–1860.
- Martin, A.R.; Chartoire, A.; Slawin, A.M.Z.; Nolan, S.P. Extending the utility of [Pd(NHC)(cinnamyl)Cl] precatalysts: Direct arylation of heterocycles. Beilstein J. Org. Chem. 2012, 8, 1637–1643.
- Li, Y.; Wang, J.; Huang, M.; Wang, Z.; Wu, Y.; Wu, Y. Direct C-H arylation of thiophenes at low catalyst loading og a phosphine-free bis (alkoxo) palladium complex. J. Org. Chem. 2014, 79, 2890–2897.
- He, X.-X.; Li, Y.; Ma, B.-B.; Ke, Z.; Liu, F.-S. Sterically encumbered tetraarylimidazolium carbine Pd-PEPPSI complexes: Highly eddicient direct arylation of imidazoles with aryl bromides under aerobic conditions. Organometallics 2016, 35, 2655–2663.
- Aktaş, A.; Celepci, D.B.; Gök, Y. Nover 2-hydroxyethyl substituted N-coordinate-Pd (II) (NHC) and bis (NHC) Pd (II) complexes: Synthesis, characterization and the catalytic activity in the direct arylation reaction. J. Chem. Sci. 2019, 131, 78.
- El Abbouchi, A.; Koubachi, J.; El Brahmi, N.; El Kazzouli, S. Direct arylation and Suzuki-Miyaura coupling of imidazo [1,2-a]pyridines catalysed by (SIPr) Pd (allyl) Cl complex under microwave-irradiation. Med. J. Chem. 2019, 9, 347–354.
- Şhain, N.; Gürbüz, N.; Karbiyik, H.; Karabiyik, H.; Özdemir, İ. Arylation of heterocyclic compounds by benzimidazole-based N-heterocylic carbene-palladium (II) complexes. J. Organomet. Chem. 2020, 907, 121076.
- Kaloğlu, M.; Kaloğlu, N.; Özdemir, I. Palladium-PEPPSI-NHC complexes bearing imidazolidin-2-ylidene ligand: Efficient precatalysts for the direct C5-arylation of N-methylpyrrole-2-carboxaldehyde. Catal. Lett. 2021, 1–16.
- Sun, H.-Y.; Gorelsky, S.I.; Stuart, D.R.; Campeau, L.-C.; Fagnou, K. Mechanistic analysis of azine N-oxide direct arylation: Evidence for a critical role of acetate in the Pd (OAc)2 precatalyst. J. Org. Chem. 2010, 75, 8180–8189.
- Nakao, Y.; Kanyiva, K.S.; Oda, S.; Hiyama, T. Hydroheteroarylation of alkynes under mild nickel catalysis. J. Am. Chem. Soc. 2006, 128, 8146–8147.
- García-Cuadrado, D.; Braga, A.A.C.; Maseras, F.; Echavarren, A.M. Proton abtraction mechanism for the palladium-catalyzed intramolecular arylation. J. Am. Chem. Soc. 2006, 128, 1066–1067.
- Zhang, W.; Zeng, Q.; Zhang, X.; Tian, Y.; Yue, Y.; Guo, Y.; Wang, Z. Ligand-free CuO nanospindle catalysed arylation of heterocycle C-H bonds. J. Org. Chem. 2011, 76, 4741–4745.
- Salamanca, V.; Aléniz, A.C. Faster palladium-catalyzed arylation of simple arenes in the presence of a methylketone: Beneficial effect of an a piori interfering solvent in C-H activation. Org. Chem. Front. 2021, 8, 1941–1951.
- Lane, B.S.; Brown, M.A.; Sames, D. Direct palladium-catalyzed C-2 and C-3 arylation of indoles: A mechanistic rationale for regioselectivity. J. Am. Chem. Soc. 2005, 127, 8050–8057.
- Martín-Matute, B.; Mateo, C.; Cárdenas, D.J.; Echavarren, A.M. Intramolecular C-H activation by alkylpalladium(II) complexes: Insights into the mechanism of the palladium-catalyzed arylation reaction. Chem. Eur. J. 2001, 7, 2341–2348.
- Park, C.-H.; Ryabova, V.; Seregin, I.V.; Sromek, A.W.; Gevorgyan, V. Palladium-catalyzed arylation and heteroarylation of indolizines. Org. Lett. 2004, 6, 1159–1162.
- Zolliger, H. Hydrogen isotope effects in aromatic substitution reactions. Adv. Phys. Org. Chem. 1964, 2, 163–200.
- Gallego, D.; Baquero, E.A. Recent advances on mechanistic studies on C–H activation catalyzed by base metals. Open Chem. 2018, 16, 1001–1058.
- Davies, D.L.; Donald, S.M.; Macgregor, S.A. Computational study of the mechanism of cyclometalation by palladium acetate. J. Am. Chem. Soc. 2005, 127, 13754–13755.
- Hennessy, E.J.; Buchwald, S.L. Synthesis of substituted oxindoles from a-chloroacetanilides via palladium-catalyzed C-H functionalization. J. Am. Chem. Soc. 2003, 125, 12084–12085.
- Zhang, C.; Tang, C.; Jiao, N. Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process. Chem. Soc. Rev. 2012, 41, 3464–3484.
- Mota, A.J.; Dedieu, A.; Bour, C.; Suffert, J. Cyclocarbopalladation involving an unusual 1,5-palladium vinyl to aryl shift as termination step: Theoretical study of the mechanism. J. Am. Chem. Soc. 2005, 127, 7171–7182.
- Boeglin, D.; Cantel, S.; Heitz, A.; Martinez, J.; Fehrentz, J.-A. Solution and solid-supported synthesis of 3,4,5-trisubstituted 1,2,4-triazole-based peptidomimetics. Org. Lett. 2003, 5, 4465–4468.
- Davies, D.-L.; Macgregor, S.A.; McMullin, C.L. Computational studies of carboxylate-assisted C-H activation and functionalization at group 8–10 transition metal centers. Chem Rev. 2017, 117, 8649–8709.
- Capito, E.; Brown, J.M.; Ricci, A. Directed palladation: Fine tuning permits the catalytic 2-alkenylation of indoles. Chem. Commun. 2005, 1854–1856.
- Hughes, C.C.; Trauner, D. Concise total synthesis of (-)-frondosin B using a novel palladium-catalyzed cyclization. Angew. Chem. Int. Ed. 2002, 41, 1569–1572.
- Campo, M.A.; Huang, Q.; Yao, T.; Tian, Q.; Larock, R.C. 1,4-Palladium migration via C-H activation, followed by arylation: Synthesis of fused polycycles. J. Am. Chem. Soc. 2003, 125, 11506–11507.
- Flegeau, E.F.; Bruneau, C.; Dixneuf, P.; Jutand, A. Autocatalysis for C-H bond activation by ruthenium (II) complexes in catalytic arylation of functional arenes. J. Am. Chem. Soc. 2011, 133, 10161–10170.
- Shan, C.; Luo, X.; Qi, X.; Liu, S.; Li, Y.; Yu Lan, Y. Mechanism of Ruthenium-Catalyzed Direct Arylation of C–H Bonds in Aromatic Amides: A Computational Study. Organometallics 2016, 35, 1440–1445.
- Campeau, L.-C.; Bertrand-Laperle, M.; Leclerc, J.-P.; Villemure, E.; Gorelsky, S.; Fagnou, K. C2, C5 and C4 azole N-oxide direct arylation including room-temperature reactions. J. Am. Chem. Soc. 2008, 130, 3276–3277.
- Rousseau, G.; Breit, B. Removable directing groups in organic synthesis and catalysis. Angew. Chem. Int. Ed. 2011, 50, 2450–2494.
- Kim, J.; Jo, M.; So, W.; No, Z. Pd-catalyzed ortho-arylation of 3,4-dihydroisoquinolones via C-H bond activation: Synthesis of 8-aryl-1,2,3,4-tetrahydroisoquinolines. Tetrahedron Lett. 2009, 50, 1229–1235.
- Choi, H.; Min, M.; Peng, Q.; Kang, D.; Paton, R.S.; Hong, S. Unraveling innate substrate control in site-selective palladium-catalyzed C–H heterocycle functionalization. Chem. Sci. 2016, 7, 3900–3909.
- Tlahuext-Aca, A.; Lee, S.Y.; Sakamoto, S.; Hartwig, J.F. Direct arylation of simple arenes with aryl bromides by synergistic silver and palladium catalysis. ACS Catal. 2021, 11, 1430–1434.
- Moroni, F.; Cozzi, A.; Chiaguri, A.; Formentini, L.; Camaioni, E.; Pellegrini-Giampietro, D.E.; Chen, Y.; Liang, S.; Zaleska, M.M.; Gonzales, C.; et al. Long-lasting neuroprotection and neurological improvement in stroke models with new, potent and brain permeable inhibitors of poly (ADP-ribose) polymerase. Br. J. Pharma. 2012, 165, 1487–1500.
- Hegan, D.C.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly (ADP-ribose) polymerase down-regulates BRCA1 and RAD51 in a pathway mediated by E2F4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206.
- Aoyama, H.; Sugita, K.; Nakamura, M.; Aoyama, A.; Salim, T.A.M.; Okamoto, M.; Baba, M.; Hashimoto, Y. Fused heterocyclic amido compounds as anti-hepatitis C virus agents. Bioorg. Med. Chem. 2011, 19, 2675–2687.
- Dyker, G. Handbook of C-H Transformations, 1st ed.; Wiley-VCH: Weinheim, Germany, 2005.
- Deshpande, P.P.; Martin, O.R. A concise total synthesis of the aglycone of the gilvocarcins. Tetrahedron Lett. 1990, 44, 6313–6316.
- Hemberge, Y.; Zhang, G.; Brun, R.; Kaiser, M.; Bringmann, G. Highly antiplasmodial non-natural oxidative products of dioncophylline A: Synthesis, absolute configuration, and conformational stability. Chem. Eur. J. 2015, 21, 14507–14518.
- Bringmann, G.; Walter, R.; Weirich, R. The Directed synthesis of biaryl compounds: Modern concepts and strategies. Angew. Chem. Int. Ed. 1990, 29, 977–991.
- Bringmann, G.; Pabst, T.; Henschel, P.; Kraus, J.; Peters, K.; Peters, E.-M.; Rycroft, D.S.; Connolly, J.D. Nondynamic and dynamic kinetic resolution of lactones with stereogenic centers and axes: Stereoselective total synthesis of herbertenediol and mastigophorenes A and B. J. Am. Chem. Soc. 2000, 122, 9127–9133.
- Harayama, T.; Yasuda, H.; Akiyama, T.; Takeuchi, Y.; Abe, H. Synthesis of arnottin I through a palladium-mediated aryl-aryl coupling reaction. Chem. Pharm. Bull. 2000, 48, 861–864.
- Hermann, W.A.; Brossmer, C.; Reisinger, C.-P.; Riermeier, T.H.; Öfele, K.; Beller, M. Palladacycles: Efficient new catalyst for the heck vinylation of aryl halides. Chem. Eur. J. 1997, 8, 1357–1364.
- Bringmann, G.; Ochse, M.; Götz, R. First atropo-divergent total synthesis of the antimalarial korupensamines A and B by the “lactone method”. J. Org. Chem. 2000, 65, 2069–2077.
- Rice, J.E.; Cai, Z.-W.; He, Z.-M.; LaVoie, E.J. Some observations on the palladium-catalyzed triflate-arene cyclization of electron-rich biaryl substrates. J. Org. Chem. 1995, 24, 8101–8104.
- Wang, L.; Shevlin, P.B. Formation of benzo[ghi]fluoranthenes by palladium catalyzed intramolecular coupling. Tetrahedron Lett. 2000, 41, 285–288.
- Echavarren, A.M.; Gómez-Lor, B.; González, J.J.; de Frutos, O. Palladium-catalyzed intramolecular arylation reaction: Mechanism and application for the synthesis of polyarenes. Synlett 2003, 2003, 585–597.
- Moulton, C.J.; Shaw, B.L. Transition metal-carbon bonds. Part XLII. Complexes of nickel, palladium, platinum, rhodium and iridium with the tridentate ligand 2,6-bis[(di-t-butylphosphino)methyl]phenyl. J. Chem. Soc. Dalton Trans. 1976, 1020–1024.
- Morales-Morales, D.; Jensen, C.M. The Chemistry of Pincer Compounds, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2007.
- Szabó, K.J.; Wendt, O.F. Pincer and Pincer-Type Complexes, 1st ed.; Wiley-VCH: Weinheim, Germany, 2014.
- Singleton, J.T. The use of pincer complexes in organic synthesis. Tetrahedron 2003, 59, 1837–1857.
- Morales-Morales, D.; Redón, R.; Yung, C.; Jensen, C.M. Dehydrogenation of alkanes catalyzed by an iridium phosphinito PCP pincer complex. Inorg. Chimica Acta 2004, 357, 2953–2956.
- Hao, X.-Q.; Wang, Y.-N.; Liu, J.-R.; Wang, K.-L.; Gong, J.-F.; Song, M.-P. Unsymmetrical, oxazolinyl-containing achiral and chiral NCN pincer ligand precursors and their complexes with palladium (II). J. Organomet. Chem. 2010, 695, 82–89.
- Liu, N.; Li, X.; Sun, H. Synthesis and properties of novel ortho-metalated cobalt (I) and iron (II) complexes through Csp2-H bond activation of dibenzylphenylphosphine. J. Organomet. Chem. 2011, 696, 2537–2542.
- Gunanathan, C.; Milstein, D. Bond activation and catalysis by ruthenium pincer complexes. Chem. Rev. 2014, 114, 12024–12087.
- Shih, W.-C.; Gu, W.; Macinnis, M.C.; Herbert, D.E.; Ozerov, O.V. Bory/borane interconversion and diversity of binding modes of oxygenous ligands in PBP pincer complexes of rhodium. Organometallics 2017, 36, 1718–1726.
- Mukherjee, A.; Milstein, D. Homogeneous catalysis by cobalt and manganese pincer complexes. ACS Catal. 2018, 8, 11435–11469.
- Churruca, F.; SanMartin, R.; Tellitu, I.; Domínguez, E. N-heterocyclic NCN-pincer palladium complexes: A source for general, highly efficient catalysts in Heck, Suzuki, and Sonogashira coupling reactions. Synlett 2005, 2005, 3116–3120.
- Takenaka, K.; Uozumi, Y. Development of chiral pincer palladium complexes bearing a pyrroloimidazolone unit. Catalytic use for asymmetric Michael addition. Org. Lett. 2004, 6, 1833–1835.
- Sun, Y.; Koehler, C.; Tan, R.; Annibale, V.T.; Song, D. Ester hydrogenation catalysed by Ru-CNN pincer complexes. Chem. Commun. 2011, 47, 8349–8351.
- Serra, D.; Cao, P.; Cabrera, J.; Padilla, R.; Rominger, F.; Limbach, M. Develepment of platinum(I) and –(IV) CNC pincer complexes and their application in a hydrovinylation reaction. Organometallics 2011, 30, 1885–1895.
- Moure, M.J.; SanMartin, R.; Domínguez, E. Copper pincer complexes as advantageous catalysts for the heteroannulation of ortho-halophenols and alkynes. Adv. Synth. Catal. 2011, 356, 2070–2080.
- Kim, D.; Le, L.; Drance, M.J.; Jensen, K.H.; Bofdanovski, K.; Cervarich, T.N.; Barnard, M.G.; Pudalov, N.J.; Knapp, S.M.M.; Chianese, A.R. Ester hydrogenation catalysed by CNN-pincer complexes of ruthenium. Organometallics 2016, 35, 982–989.
- Urgoitia, G.; SanMartin, R.; Herrero, M.T.; Domínguez, E. Efficient copper-free aerobic alkyne homocoupling in polyethylene glycol. Environ. Chem. Lett. 2017, 15, 157–164.
- Gorgas, N.; Alves, L.G.; Stöger, B.; Martins, A.M.; Veiros, L.F.; Kirchner, K. Stable, yet highly reactive nonclassical iraon(II) polyhydide pincer complexes: Z-selective dimerization and hydroboration of terminal alkynes. J. Am. Chem. Soc. 2017, 139, 8130–8133.
- González-Sebastián, L.; Morales-Morales, D. Cross-coupling reactions catalysed by palladium pincer complexes. A review of recent advances. J. Organomet. Chem. 2019, 893, 39–51.
- Churruca, F.; Hernández, S.; Perea, M.; SanMartin, R.; Domínguez, E. Direct Access to pyrazolo(benzo)thienoquinolines. Highly effective palladium catalysts for the intramolecular C-H heteroarylation of arenes. Chem. Commun. 2013, 49, 1413–1415.
- Khake, S.M.; Soni, V.; Gonnade, R.G.; Punji, B. Design and development of POCN-pincer palladium catalysts for C-H bond arylation of azoles with aryl iodides. Dalton Trans. 2014, 43, 16084–16096.
- Khake, S.M.; Jagtap, R.A.; Dangat, Y.B.; Gonnade, R.G.; Vanka, K.; Punji, B. Mechanistic insights into pincer-ligated palladium-catalyzed arylation of azoles with aryl iodides: Evidence of a PdII-PdIV-PdII pathway. Organometallics 2016, 35, 875–886.
- Conde, N.; Churruca, F.; SanMartin, R.; Herrero, M.T.; Domínguez, E. A further decrease in the catalyst loading for the palladium-catalyzed direct intramolecular arylation of amides and sulphonamides. Adv. Synth. Catal. 2015, 357, 1525–1531.
- Benaglia, M. Recoverable and Recyclable Catalysts; John Wiley & Sons: Chippenham, UK, 2009.
- Wang, C.; Li, Y.; Lu, B.; Hao, X.-Q.; Gong, J.-F.; Song, M.-P. (Phosphinito)aryl benzimidazole PCN pincer palladium (II) complexes: Synthesis, characterization and catalytic activity in C-H arylation of azoles with aryl iodides. Polyhedron 2018, 143, 184–192.
- Öfele, K. 1,3-dimethyl-4-imidazolinyliden-(2)-pentacarbonylchrom ein neuer übergangsmetall-carben-komplex. J. Organomet. Chem. 1968, 12, P42–P43.
- Lee, J.-Y.; Lee, J.-Y.; Chang, Y.-Y.; Hu, C.-H.; Wang, N.M.; Lee, H.M. Palladium complexes with tridentate N-heterocyclic carbine ligands: Selective “normal” and “abnormal” binding and thir anticancer activities. Organometallics 2015, 34, 4359–4368.
- Corberán, R.; Mas-Marzá, E.; Peris, E. Mono-, bi- and tridentate N-heterocyclic carbene ligands for the preparation of transition-metal-based homogeneous catalysts. Eur. J. Inorg. Chem. 2009, 2009, 1700–1716.
- Bhaskar, R.; Sharma, A.K.; Singh, A.K. Palladium (II) complexes of N-heterocyclic carbine amidates derived from chalcogenated acetamide-functionalized 1H-benzimidazolium salts: Recyclable catalyst for regioselective arylation of imidazoles under aerobic conditions. Organometallics 2018, 37, 2669–2681.
- Bhatt, R.; Bhuvanesh, N.; Sharma, K.N.; Joshi, H. Palladium complexes of thio/seleno-ether containing N-heterocyclic carbenes: Efficient and reusable catalyst for regioselective C-H bond arylation. Eur. J. Inorg. Chem. 2020, 2020, 532–540.
- Widegren, J.A.; Finke, R.G. A review of the problem of distinguishing true homogeneous catalysis from soluble or other metal-particle heterogeneous catalysis under reducing conditions. J. Mol. Catal. A Chem. 2003, 198, 317–341.
- Lee, J.-Y.; Shen, J.-S.; Tzeng, R.-J.; Lu, I.-C.; Lii, J.-H.; Hu, C.-H.; Lee, H.M. Well-defined palladium (0) complexes bearing N-heterocyclic carbene and phosphine moieties: Efficient catalytic applications in Mizoroki-Heck reaction and direct C-H fuctionalization. Dalton Trans. 2016, 45, 10375–10388.
- Chartoire, A.; Frogneux, X.; Boreux, A.; Slawin, A.M.Z.; Nolan, S.P. [Pd(IPr*)(3-Cl-pyridinyl)Cl2]: A novel and efficient PEPPSI precatalyst. Organometallics 2012, 31, 6947–6951.
- Li, H.-H.; Maitra, R.; Kuo, Y.-T.; Chen, J.-H.; Hu, C.-H.; Lee, H.M. A tridentate CNO-donor palladium (II) complex as a efficient catalyst for direct C-H arylation: Application in preparation of imidazole-based push-pull chromophores. Appl. Organomet. Chem. 2018, 32, 3956.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.6K
Entry Collection:
Organic Synthesis
Revisions:
2 times
(View History)
Update Date:
09 Aug 2021
Table of Contents


Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No