 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Teresa Frisan | + 4430 word(s) | 4430 | 2021-07-09 08:53:51 | | | |
| 2 | Catherine Yang | -5 word(s) | 4425 | 2021-08-05 12:05:34 | | |
Video Upload Options
The idea that bacterial toxins are not only killers but also execute more sophisticated roles during bacteria–host interactions by acting as negotiators has been highlighted in the past decades. Depending on the toxin, its cellular target and mode of action, the final regulatory outcome can be different. Focused on two families of bacterial toxins: genotoxins and pore-forming toxins, which have different modes of action but share the ability to modulate the host’s immune responses, independently of their capacity to directly kill immune cells.
1. Introduction
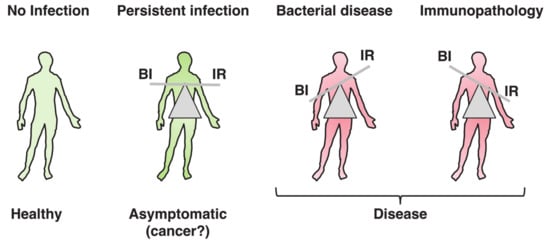
The word toxin according to the definition by Merriam-Webster is “a poisonous substance that is a specific product of the metabolic activities of a living organism and is usually very unstable, notably toxic when introduced into tissues, and typically capable of inducing antibody formation,” which entails the concept of tissue destruction (https://www.merriam-webster.com/dictionary/toxin#note-1). Results from the past decades have highlighted the possibility that the biological function of bacterial toxins, key virulence factors in bacterial pathogenesis [1][2], may not exclusively be to cause damage to the host tissue, allowing bacteria to colonize, invade, and spread. When this delicate equilibrium is broken, due to high toxin (bacteria)-induced tissue damage or uncontrolled immune responses, pathological conditions develop, as summarized in Figure 1. The killer vs. negotiator paradox is directly connected to another highly relevant issue in bacterial pathogenesis, namely the amount of toxin that is produced during the course of an in vivo infection.
In this review, we will focus on the immunomodulatory role of two families of bacterial toxins: (a) genotoxins (BTGXs) and (b) pore-forming toxins (PFTs). We have chosen these two effectors, among all the bacterial exotoxins, which may have similar outcomes on the regulation of the host response because they have very different modes of action and target the two innermost and outermost essential structures of a cell, respectively: the DNA and the plasma membrane; the alpha and omega. However, as immunomodulators, they can achieve very similar outcomes, often targeting common node regulators such as transcription factors (e.g., NFκB) and protein kinases, highlighting the relevance of their activities during host–pathogen interactions (Figure 2). For both toxin families, the best-characterised immunomodulatory effect is promotion of proinflammatory responses [2][3].

BTGXs induce DNA damage in target host cells [4]. The subsequent cellular response is characterised by secretion of a wide variety of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, and TNFα) within a period of 5 to 72 h post-intoxication in vitro [3], possibly via NFκB activation induced by the cellular DNA damage response (DDR) pathway [5]. However, there is also a late pro-inflammatory effect (from 4 days post-intoxication onwards), associated with induction of senescence, a permanent quiescent status that prevents growth of DNA-damaged cells. Senescent cells secrete a highly pro-inflammatory secretome, known as senescence-associated secretory phenotype (SASP) [6][7].
The pro-inflammatory response mediated by PFTs is linked to their capacity to: (i) induce necrosis or pyroptosis, and the subsequent release of damage-associated molecular pattern (DAMPs) into the extracellular environment [8][9]; (ii) activation of the NLR family pyrin domain-containing 3 (NALP3) inflammasome through diverse mechanisms such as alteration of the ion homeostasis (efflux of potassium K+and influx of calcium Ca2+), or rupture of the phagosome/lysosome compartments, resulting in release of the pro-inflammatory cytokines IL1β and IL-18 [10]; (iii) activation of the transcription factor NFκB via induction of Ca2+oscillation at sublethal doses, leading to transcriptional activation and secretion of a broad panel of pro-inflammatory cytokines, such as IL-6 and IL-8 [11][12].
However, another interesting aspect of the immunomodulatory effects of PFTs and BTGXs is their anti-inflammatory activity, which goes beyond their capacity to induce cell death in general or specifically in cells of the innate or adaptive immune system [3][13][14]. This outcome relies on the manipulation of the immune response, resulting in: (i) suppression of pro-inflammatory cytokines, (ii) re-polarization of the immune response, and (iii) balance between toxin production and bacterial fitness Thus, these effectors can take control and modulate the mechanisms of host tolerance to infection, defined as “reduction of the negative impact of infection on host fitness without affecting the pathogen burden” [15] to their own advantage.
2. Bacterial Genotoxins
2.1. General Overview and Mode of Action
2.1.1. Cytolethal Distending Toxins
2.1.2. Typhoid Toxin
2.1.3. Colibactin
2.1.4. DNA Damage Response
2.2. Biological Functions of BTGXs
2.2.1. Unresolved DNA Damage Induced by BTGXs
Apoptosis Induced by Bacterial Genotoxins
BTGXs-Induced Senescence
2.2.2. Acquisition of Genomic Instability
2.2.3. Immunomodulatory Properties of BTGX
References
- Frisan, T.; Guidi, R.; Guerra, L. Toxins acting on intracellular targets: Only foes or also friends? In Bacterial Pathogenesis: Molecular and Cellular Mechanisms; Locht, C., Simonet, S., Eds.; Caister Academic Press: Norfolk, UK, 2012.
- Los, F.C.O.; Randis, T.M.; Aroian, R.V.; Ratner, A.J. Role of Pore-Forming Toxins in Bacterial Infectious Diseases. Microbiol. Mol. Biol. Rev. 2013, 77, 173–207.
- Martin, O.C.B.; Frisan, T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins 2020, 12, 63.
- Grasso, F.; Frisan, T. Bacterial Genotoxins: Merging the DNA Damage Response into Infection Biology. Biomolecules 2015, 5, 1762–1782.
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326.
- He, S.H.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011.
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827.
- Bischofberger, M.; Iacovache, I.; van der Goot, F.G. Pathogenic Pore-Forming Proteins: Function and Host Response. Cell Host Microbe 2012, 12, 266–275.
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release mechanisms of major DAMPs. Apoptosis 2021, 26, 152–162.
- Jing, W.D.; Lo Pilato, J.; Kay, C.; Man, S.M. Activation mechanisms of inflammasomes by bacterial toxins. Cell. Microbiol. 2021, 23, e13309.
- Soderblom, T.; Laestadius, A.; Oxhamre, C.; Aperia, A.; Richter-Dahlfors, A. Toxin-Induced calcium oscillations: A novel strategy to affect gene regulation in target cells. Int. J. Med. Microbiol. 2002, 291, 511–515.
- Nhieu, G.T.; Clair, C.; Grompone, G.; Sansonetti, P. Calcium signalling during cell interactions with bacterial pathogens. Biol. Cell 2004, 96, 93–101.
- Spaan, A.N.; van Strijp, J.A.G.; Torres, V.J. Leukocidins: Staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 2017, 15, 435–447.
- Tromp, A.T.; van Strijp, J.A.G. Studying Staphylococcal Leukocidins: A Challenging Endeavor. Front. Microbiol. 2020, 11, 611.
- Medzhitov, R.; Schneider, D.S.; Soares, M.P. Disease Tolerance as a Defense Strategy. Science 2012, 335, 936–941.
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon Off. J. Int. Soc. Toxinology 2001, 39, 1729–1736.
- Jinadasa, R.N.; Bloom, S.E.; Weiss, R.S.; Duhamel, G.E. Cytolethal distending toxin: A conserved bacterial genotoxin that blocks cell cycle progression, leading to apoptosis of a broad range of mammalian cell lineages. Microbiology 2011, 1577, 1851–1875.
- Nesic, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433.
- Elwell, C.A.; Dreyfus, L.A. DNAase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963.
- Lara-Tejero, M.; Galan, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357.
- Frisan, T.; Cortes-Bratti, X.; Chaves-Olarte, E.; Stenerlöw, B.; Thelestam, M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707.
- Fowler, C.C.; Chang, S.J.; Gao, X.; Geiger, T.; Stack, G.; Galan, J.E. Emerging insights into the biology of typhoid toxin. Curr. Opin. Microbiol. 2017, 35, 70–77.
- Fais, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More Than a New Bacterial Toxin. Toxins 2018, 10, 151.
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlow, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell Microbiol. 2005, 7, 921–934.
- Boesze-Battaglia, K.; Besack, D.; McKay, T.; Zekavat, A.; Otis, L.; Jordan-Sciutto, K.; Shenker, B.J. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal-distending toxin. Cell Microbiol. 2006, 8, 823–836.
- Lin, C.D.; Lai, C.K.; Lin, Y.H.; Hsieh, J.T.; Sing, Y.T.; Chang, Y.C.; Chen, K.C.; Wang, W.C.; Su, H.L.; Lai, C.H. Cholesterol Depletion Reduces Entry of Campylobacter jejuni Cytolethal Distending Toxin and Attenuates Intoxication of Host Cells. Infect. Immun. 2011, 79, 3563–3575.
- McSweeney, L.A.; Dreyfus, L.A. Carbohydrate-Binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060.
- Frisan, T. Bacterial genotoxins: The long journey to the nucleus of mammalian cells. Biochim. Biophys. Acta 2015, 1858, 567–575.
- Gargi, A.; Tamilselvam, B.; Powers, B.; Prouty, M.G.; Lincecum, T.; Eshraghi, A.; Maldonado-Arocho, F.J.; Wilson, B.A.; Bradley, K.A.; Blanke, S.R. Cellular interactions of the cytolethal distending toxins from Escherichia coli and Haemophilus ducreyi. J. Biol. Chem. 2013, 288, 7492–7505.
- Boesze-Battaglia, K.; Dhingra, A.; Walker, L.M.; Zekavat, A.; Shenker, B.J. Internalization and Intoxication of Human Macrophages by the Active Subunit of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin Is Dependent Upon Cellugyrin (Synaptogyrin-2). Front. Immunol. 2020, 11, 1262.
- Chen, M.X.; Chen, Y.; Fu, R.; Mao, G.Q.; Liu, S.Y.; Shen, T.B. Rab5a Promotes Cytolethal Distending Toxin B Induced Cytotoxicity and Inflammation. Infect. Immun. 2020, 88.
- Eshraghi, A.; Dixon, S.D.; Tamilselvam, B.; Kim, E.J.; Gargi, A.; Kulik, J.C.; Damoiseaux, R.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxins require components of the ER-associated degradation pathway for host cell entry. PLoS Pathog. 2014, 10, e1004295.
- Spano, S.; Ugalde, J.E.; Galan, J.E. Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment. Cell Host Microbe 2008, 3, 30–38.
- Song, J.; Gao, X.; Galan, J.E. Structure and function of the Salmonella Typhi chimaeric A(2)B(5) typhoid toxin. Nature 2013, 499, 350–354.
- Desai, P.T.; Porwollik, S.; Long, F.; Cheng, P.; Wollam, A.; Clifton, S.W.; Weinstock, G.M.; McClelland, M. Evolutionary Genomics of Salmonella enterica Subspecies. mBio 2013, 4.
- Suez, J.; Porwollik, S.; Dagan, A.; Marzel, A.; Schorr, Y.I.; Desai, P.T.; Agmon, V.; McClelland, M.; Rahav, G.; Gal-Mor, O. Virulence Gene Profiling and Pathogenicity Characterization of Non-Typhoidal Salmonella Accounted for Invasive Disease in Humans. PLoS ONE 2013, 8, e58449.
- Rodriguez-Rivera, L.D.; Bowen, B.M.; den Bakker, H.C.; Duhamel, G.E.; Wiedmann, M. Characterization of the cytolethal distending toxin (typhoid toxin) in non-typhoidal Salmonella serovars. Gut Pathog. 2015, 7, 1–7.
- Deng, L.Q.; Song, J.M.; Gao, X.; Wang, J.W.; Yu, H.; Chen, X.; Varki, N.; Naito-Matsui, Y.; Galan, J.E.; Varki, A. Host Adaptation of a Bacterial Toxin from the Human Pathogen Salmonella Typhi. Cell 2014, 159, 1290–1299.
- Chang, S.J.; Jin, S.C.; Jiao, X.Y.; Galan, J.E. Unique features in the intracellular transport of typhoid toxin revealed by a genome-wide screen. PLoS Pathog. 2019, 15, e1007704.
- Guidi, R.; Levi, L.; Rouf, S.F.; Puiac, S.; Rhen, M.; Frisan, T. Salmonella enterica delivers its genotoxin through outer membrane vesicles secreted from infected cells. Cell. Microbiol. 2013, 15, 2034–2050.
- Suresh, A.; Shaik, S.; Baddam, R.; Ranjan, A.; Qumar, S.; Jadhav, S.; Semmler, T.; Ghazi, I.A.; Wieler, L.H.; Ahmed, N. Evolutionary Dynamics Based on Comparative Genomics of Pathogenic Escherichia coli Lineages Harboring Polyketide Synthase (pks) Island. mBio 2021, 12, e03634-20.
- Nougayrede, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851.
- Xue, M.Z.; Kim, C.S.; Healy, A.R.; Wernke, K.M.; Wang, Z.X.; Frischling, M.C.; Shine, E.E.; Wang, W.W.; Herzon, S.B.; Crawford, J.M. Structure elucidation of colibactin and its DNA cross-links. Science 2019, 365, eaax2685.
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrede, J.P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. mBio 2018, 9, e02393-17.
- Healy, A.R.; Nikolayevskiy, H.; Patel, J.R.; Crawford, J.M.; Herzon, S.B. A Mechanistic Model for Colibactin-Induced Genotoxicity. J. Am. Chem. Soc. 2016, 138, 15563–15570.
- Tripathi, P.; Shine, E.E.; Healy, A.R.; Kim, C.S.; Herzon, S.B.; Bruner, S.D.; Crawford, J.M. ClbS Is a Cyclopropane Hydrolase That Confers Colibactin Resistance. J. Am. Chem. Soc. 2017, 139, 17719–17722.
- Reuter, C.; Alzheimer, M.; Walles, H.; Oelschlaeger, T.A. An adherent mucus layer attenuates the genotoxic effect of colibactin. Cell. Microbiol. 2018, 20, e12812.
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817.
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kacmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85.
- Walden, H.; Deans, A.J. The Fanconi anemia DNA repair pathway: Structural and functional insights into a complex disorder. Annu. Rev. Biophys. 2014, 43, 257–278.
- Kitagawa, T.; Hoshida, H.; Akada, R. Genome-Wide analysis of cellular response to bacterial genotoxin CdtB in yeast. Infect. Immun. 2007, 75, 1393–1402.
- Fedor, Y.; Vignard, J.; Nicolau-Travers, M.L.; Boutet-Robinet, E.; Watrin, C.; Salles, B.; Mirey, G. From single-strand breaks to double-strand breaks during S-phase: A new mode of action of the Escherichia coli Cytolethal Distending Toxin. Cell. Microbiol. 2013, 15, 1–15.
- Fahrer, J.; Huelsenbeck, J.; Jaurich, H.; Dorsam, B.; Frisan, T.; Eich, M.; Roos, W.P.; Kaina, B.; Fritz, G. Cytolethal distending toxin (CDT) is a radiomimetic agent and induces persistent levels of DNA double-strand breaks in human fibroblasts. DNA Repair 2014, 18, 31–43.
- Pucci, B.; Kasten, M.; Giordano, A. Cell cycle and apoptosis. Neoplasia 2000, 2, 291–299.
- Green, D.R.; Llambi, F. Cell Death Signaling. CSH Perspect. Biol. 2015, 7, a006080.
- Shenker, B.J.; McKay, T.; Datar, S.; Miller, M.; Chowhan, R.; Demuth, D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 1999, 162, 4773–4780.
- Shenker, B.J.; Hoffmaster, R.H.; Zekavat, A.; Yamaguchi, N.; Lally, E.T.; Demuth, D.R. Induction of apoptosis in human T cells by Actinobacillus actinomycetemcomitans cytolethal distending toxin is a consequence of G2 arrest of the cell cycle. J. Immunol. 2001, 167, 435–441.
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Miyauchi, M.; Takata, T.; Sugai, M. Caspase-2 and caspase-7 are involved in cytolethal distending toxin-induced apoptosis in Jurkat and MOLT-4 T-cell lines. Infect. Immun. 2004, 72, 871–879.
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Nakachi, K.; Fujiwara, T.; Komatsuzawa, H.; Sugai, M. Cytolethal distending toxin induces caspase-dependent and -independent cell death in MOLT-4 cells. Infect. Immun. 2008, 76, 4783–4791.
- Svensson, L.; Tarkowski, A.; Thelestam, M.; Lagergård, T. The impact of Haemophilus ducreyi cytolethal distending toxin on cells involved in immune response. Microb. Pathog. 2001, 30, 157–166.
- Wising, C.; Azem, J.; Zetterberg, M.; Svensson, L.A.; Ahlman, K.; Lagergard, T. Induction of apoptosis/necrosis in various human cell lineages by Haemophilus ducreyi cytolethal distending toxin. Toxicon Off. J. Int. Soc. Toxinol. 2005, 45, 767–776.
- Li, G.; Niu, H.; Zhang, Y.H.; Li, Y.L.; Xie, F.; Langford, P.R.; Liu, S.; Wang, C.L. Haemophilus parasuis cytolethal distending toxin induces cell cycle arrest and p53-dependent apoptosis. PLoS ONE 2017, 12, e0177199.
- Shenker, B.J.; Boesze-Battaglia, K.; Scuron, M.D.; Walker, L.P.; Zekavat, A.; Dlakic, M. The toxicity of the Aggregatibacter actinomycetemcomitans cytolethal distending toxin correlates with its phosphatidylinositol-3,4,5-triphosphate phosphatase activity. Cell. Microbiol. 2016, 18, 223–243.
- Shenker, B.J.; Walker, L.M.; Zekavat, A.; Weiss, R.H.; Boesze-Battaglia, K. The Cell-Cycle Regulatory Protein p21(CIP1/WAF1) Is Required for Cytolethal Distending Toxin (Cdt)-Induced Apoptosis. Pathogens 2020, 9, 38.
- Liyanage, N.P.M.; Manthey, K.C.; Dassanayake, R.P.; Kuszynski, C.A.; Oakley, G.G.; Duhamel, G.E. Helicobacter hepaticus Cytolethal Distending Toxin Causes Cell Death in Intestinal Epithelial Cells via Mitochondrial Apoptotic Pathway. Helicobacter 2010, 15, 98–107.
- Humphreys, D.; ElGhazaly, M.; Frisan, T. Senescence and Host–Pathogen Interactions. Cells 2020, 9, 1747.
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial Intoxication Evokes Cellular Senescence with Persistent DNA Damage and Cytokine Signaling. J. Cell Mol. Med. 2010, 14, 357–367.
- Ibler, A.E.M.; Elghazaly, M.; Naylor, K.L.; Bulgakova, N.A.; El-Khamisy, S.F.; Humphreys, D. Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat. Commun. 2019, 10, 4040.
- Secher, T.; Samba-Louaka, A.; Oswald, E.; Nougayrede, J.P. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS ONE 2013, 8, e77157.
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Dechelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942.
- Dalmasso, G.; Cougnoux, A.; Delmas, J.; Darfeuille-Michaud, A.; Bonnet, R. The bacterial genotoxin colibactin promotes colon tumor growth by modifying the tumor microenvironment. Gut Microbes 2014, 5, 675–680.
- Pere-Vedrenne, C.; Prochazkova-Carlotti, M.; Rousseau, B.; He, W.; Chambonnier, L.; Sifre, E.; Buissonniere, A.; Dubus, P.; Megraud, F.; Varon, C.; et al. The Cytolethal Distending Toxin Subunit CdtB of Helicobacter hepaticus Promotes Senescence and Endoreplication in Xenograft Mouse Models of Hepatic and Intestinal Cell Lines. Front. Cell. Infect. Microbiol. 2017, 7, 268.
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674.
- Guidi, R.; Guerra, L.; Levi, L.; Stenerlow, B.; Fox, J.G.; Josenhans, C.; Masucci, M.G.; Frisan, T. Chronic exposure to the cytolethal distending toxins of Gram-negative bacteria promotes genomic instability and altered DNA damage response. Cell. Microbiol. 2013, 15, 98–113.
- Cuevas-Ramos, G.; Petit, C.R.; Marcq, I.; Boury, M.; Oswald, E.; Nougayrede, J.P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc. Natl. Acad. Sci. USA 2010, 107, 11537–11542.
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473.
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12, 1–15.
- Muzny, D.M.; Bainbridge, M.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Armaghany, T.; Wilson, J.D.; Chu, Q.; Mills, G. Genetic alterations in colorectal cancer. Gastrointest. Cancer Res. 2012, 5, 19–27.
- Kiesler, P.; Fuss, I.J.; Strober, W. Experimental Models of Inflammatory Bowel Diseases. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 154–170.
- Shen, Z.; Feng, Y.; Rogers, A.B.; Rickman, B.; Whary, M.T.; Xu, S.; Clapp, K.M.; Boutin, S.R.; Fox, J.G. Cytolethal distending toxin promotes Helicobacter cinaedi-associated typhlocolitis in interleukin-10-deficient mice. Infect. Immun. 2009, 77, 2508–2516.
- Ge, Z.; Feng, Y.; Ge, L.; Parry, N.; Muthupalani, S.; Fox, J.G. Helicobacter hepaticus cytolethal distending toxin promotes intestinal carcinogenesis in 129Rag2-deficient mice. Cell. Microbiol. 2017, 19.
- Ge, Z.; Feng, Y.; Whary, M.T.; Nambiar, P.R.; Xu, S.; Ng, V.; Taylor, N.S.; Fox, J.G. Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect. Immun. 2005, 73, 3559–3567.
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123.
- Arthur, J.C.; Gharaibeh, R.Z.; Muhlbauer, M.; Perez-Chanona, E.; Uronis, J.M.; McCafferty, J.; Fodor, A.A.; Jobin, C. Microbial genomic analysis reveals the essential role of inflammation in bacteria-induced colorectal cancer. Nat. Commun. 2014, 5, 1–11.
- Dejea, C.M.; Wick, E.C.; Hechenbleikner, E.M.; White, J.R.; Welch, J.L.M.; Rossetti, B.J.; Peterson, S.N.; Snesrud, E.C.; Borisy, G.G.; Lazarev, M.; et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc. Natl. Acad. Sci. USA 2014, 111, 18321–18326.
- Drewes, J.L.; White, J.R.; Dejea, C.M.; Fathi, P.; Iyadorai, T.; Vadivelu, J.; Roslani, A.C.; Wick, E.C.; Mongodin, E.F.; Loke, M.F.; et al. High-Resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes 2017, 3, 34.
- Dejea, C.M.; Fathi, P.; Craig, J.M.; Boleij, A.; Taddese, R.; Geis, A.L.; Wu, X.; Shields, C.E.D.; Hechenbleikner, E.M.; Huso, D.L.; et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 2018, 359, 592–597.
- Eklof, V.; Lofgren-Burstrom, A.; Zingmark, C.; Edin, S.; Larsson, P.; Karling, P.; Alexeyev, O.; Rutegard, J.; Wikberg, M.L.; Palmqvist, R. Cancer-Associated fecal microbial markers in colorectal cancer detection. Int. J. Cancer 2017, 141, 2528–2536.
- Warren, R.L.; Freeman, D.J.; Pleasance, S.; Watson, P.; Moore, R.A.; Cochrane, K.; Allen-Vercoe, E.; Holt, R.A. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome 2013, 1, 16.
- Allali, I.; Delgado, S.; Marron, P.I.; Astudillo, A.; Yeh, J.J.; Ghazal, H.; Amzazi, S.; Keku, T.; Azcarate-Peril, M.A. Gut microbiome compositional and functional differences between tumor and non-tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes 2015, 6, 161–172.
- He, Z.; Gharaibeh, R.; Newsome, R.; Pope, J.; Dougherty, M.; Tomkovich, S.; Pons, B.; Mirey, G.J.V.; Hendrixson, D.; Vignard, J.; et al. Campylobacterjejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut 2018.
- Pere-Vedrenne, C.; Cardinaud, B.; Varon, C.; Mocan, I.; Buissonniere, A.; Izotte, J.; Megraud, F.; Menard, A. The Cytolethal Distending Toxin Subunit CdtB of Helicobacter Induces a Th17-related and Antimicrobial Signature in Intestinal and Hepatic Cells In Vitro. J. Infect. Dis. 2016, 213, 1979–1989.
- Akifusa, S.; Poole, S.; Lewthwaite, J.; Henderson, B.; Nair, S.P. Recombinant Actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect. Immun. 2001, 69, 5925–5930.
- Shenker, B.J.; Ojcius, D.M.; Walker, L.P.; Zekavat, A.; Scuron, M.D.; Boesze-Battaglia, K. Aggregatibacter actinomycetemcomitans cytolethal distending toxin activates the NLRP3 inflammasome in human macrophages, leading to the release of proinflammatory cytokines. Infect. Immun. 2015, 83, 1487–1496.
- Ando-Suguimoto, E.S.; da Silva, M.P.; Kawamoto, D.; Chen, C.; DiRienzo, J.M.; Mayer, M.P. The cytolethal distending toxin of Aggregatibacter actinomycetemcomitans inhibits macrophage phagocytosis and subverts cytokine production. Cytokine 2014, 66, 46–53.
- Belibasakis, G.N.; Johansson, A.; Wang, Y.; Chen, C.; Lagergard, T.; Kalfas, S.; Lerner, U.H. Cytokine responses of human gingival fibroblasts to Actinobacillus actinomycetemcomitans cytolethal distending toxin. Cytokine 2005, 30, 56–63.
- Hickey, T.E.; McVeigh, A.L.; Scott, D.A.; Michielutti, R.E.; Bixby, A.; Carroll, S.A.; Bourgeois, A.L.; Guerry, P. Campylobacter jejuni cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect. Immun. 2000, 68, 6535–6541.
- Zheng, J.; Meng, J.; Zhao, S.; Singh, R.; Song, W. Campylobacter-induced interleukin-8 secretion in polarized human intestinal epithelial cells requires Campylobacter-secreted cytolethal distending toxin- and Toll-like receptor-mediated activation of NF-kappaB. Infect. Immun. 2008, 76, 4498–4508.
- Fox, J.G.; Rogers, A.B.; Whary, M.T.; Ge, Z.; Taylor, N.S.; Xu, S.; Horwitz, B.H.; Erdman, S.E. Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Camplyobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect. Immun. 2004, 72, 1116–1125.
- Pratt, J.S.; Sachen, K.L.; Wood, H.D.; Eaton, K.A.; Young, V.B. Modulation of host immune responses by the cytolethal distending toxin of Helicobacter hepaticus. Infect. Immun. 2006, 74, 4496–4504.
- Young, V.B.; Knox, K.A.; Pratt, J.S.; Cortez, J.S.; Mansfield, L.S.; Rogers, A.B.; Fox, J.G.; Schauer, D.B. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 2004, 72, 2521–2527.
- Ge, Z.; Rogers, A.B.; Feng, Y.; Lee, A.; Xu, S.; Taylor, N.S.; Fox, J.G. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007, 9, 2070–2080.
- Secher, T.; Payros, D.; Brehin, C.; Boury, M.; Watrin, C.; Gillet, M.; Bernard-Cadenat, I.; Menard, S.; Theodorou, V.; Saoudi, A.; et al. Oral tolerance failure upon neonatal gut colonization with Escherichia coli producing the genotoxin colibactin. Infect. Immun. 2015, 83, 2420–2429.
- Bakthavatchalu, V.; Wert, K.J.; Feng, Y.; Mannion, A.; Ge, Z.M.; Garcia, A.; Scott, K.E.; Caron, T.J.; Madden, C.M.; Jacobsen, J.T.; et al. Cytotoxic Escherichia coli strains encoding colibactin isolated from immunocompromised mice with urosepsis and meningitis. PLoS ONE 2018, 13, e0194443.
- Guerra, L.; Carr, H.S.; Richter-Dahlfors, A.; Masucci, M.G.; Thelestam, M.; Frost, J.A.; Frisan, T. A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS ONE 2008, 3, e2254.
- Tak, P.P.; Firestein, G.S. NF-kappa B: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11.
- Cuenda, A.; Rousseau, S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim. Biophys. Acta 2007, 1773, 1358–1375.
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809.
- Ahn, J.; Gutman, D.; Saijo, S.; Barber, G.N. STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. USA 2012, 109, 19386–19391.
- Lan, Y.Y.; Londono, D.; Bouley, R.; Rooney, M.S.; Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014, 9, 180–192.
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343.
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470.
- Morrison, P.J.; Bending, D.; Fouser, L.A.; Wright, J.F.; Stockinger, B.; Cooke, A.; Kullberg, M.C. Th17-cell plasticity in Helicobacter hepaticus-induced intestinal inflammation. Mucosal Immunol. 2013, 6, 1143–1156.
- Eberl, G. Immunity by equilibrium. Nat. Rev. Immunol. 2016, 16, 524–532.
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635.
- Gelfanova, V.; Hansen, E.J.; Spinola, S.M. Cytolethal distending toxin of Haemophilus ducreyi induces apoptotic death of Jurkat T cells. Infect. Immun. 1999, 67, 6394–6402.
- Marcq, I.; Martin, P.; Payros, D.; Cuevas-Ramos, G.; Boury, M.; Watrin, C.; Nougayrede, J.P.; Olier, M.; Oswald, E. The Genotoxin Colibactin Exacerbates Lymphopenia and Decreases Survival Rate in Mice Infected With Septicemic Escherichia coli. J. Infect. Dis. 2014, 210, 285–294.
- Johnson, J.R.; Johnston, B.; Kuskowski, M.A.; Nougayrede, J.P.; Oswald, E. Molecular Epidemiology and Phylogenetic Distribution of the Escherichia coli pks Genomic Island. J. Clin. Microbiol. 2008, 46, 3906–3911.
- Olier, M.; Marcq, I.; Salvador-Cartier, C.; Secher, T.; Dobrindt, U.; Boury, M.; Bacquie, V.; Penary, M.; Gaultier, E.; Nougayrede, J.P.; et al. Genotoxicity of Escherichia coli Nissle 1917 strain cannot be dissociated from its probiotic activity. Gut Microbes 2012, 3, 501–509.
- Del Bel Belluz, L.; Guidi, R.; Pateras, I.S.; Levi, L.; Mihaljevic, B.; Rouf, S.F.; Wrande, M.; Candela, M.; Turroni, S.; Nastasi, C.; et al. The Typhoid Toxin Promotes Host Survival and the Establishment of a Persistent Asymptomatic Infection. PLoS Pathog. 2016, 12, e1005528.
- Miller, R.A.; Betteken, M.I.; Guo, X.; Altier, C.; Duhamel, G.E.; Wiedmann, M. The Typhoid Toxin Produced by the Nontyphoidal Salmonella enterica Serotype Javiana Is Required for Induction of a DNA Damage Response In Vitro and Systemic Spread In vivo. mBio 2018, 9, e00467-18.
- Martin, O.C.; Bergonzini, A.; Chiloeches, M.L.; Paparouna, E.; Butter, D.; Theodorou, S.D.; Haykal, M.M.; Boutet-Robinet, E.; Tebaldi, T.; Wakeham, A.; et al. Influence of the microenvironment on modulation of the host response by typhoid toxin. Cell Rep. 2021, 35, 108931.

