The idea that bacterial toxins are not only killers but also execute more sophisticated roles during bacteria–host interactions by acting as negotiators has been highlighted in the past decades. Depending on the toxin, its cellular target and mode of action, the final regulatory outcome can be different. FIn this review, we have focused on two families of bacterial toxins: genotoxins and pore-forming toxins, which have different modes of action but share the ability to modulate the host’s immune responses, independently of their capacity to directly kill immune cells.
1. Introduction
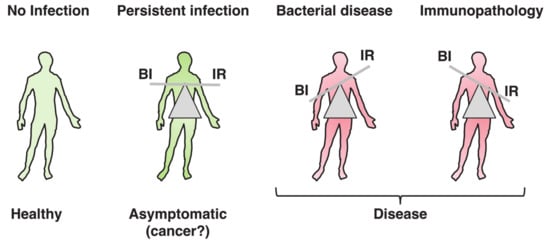
The word toxin according to the definition by Merriam-Webster is “a poisonous substance that is a specific product of the metabolic activities of a living organism and is usually very unstable, notably toxic when introduced into tissues, and typically capable of inducing antibody formation,” which entails the concept of tissue destruction (https://www.merriam-webster.com/dictionary/toxin#note-1). Results from the past decades have highlighted the possibility that the biological function of bacterial toxins, key virulence factors in bacterial pathogenesis [1][2][1,2], may not exclusively be to cause damage to the host tissue, allowing bacteria to colonize, invade, and spread. When this delicate equilibrium is broken, due to high toxin (bacteria)-induced tissue damage or uncontrolled immune responses, pathological conditions develop, as summarized in Figure 1. The killer vs. negotiator paradox is directly connected to another highly relevant issue in bacterial pathogenesis, namely the amount of toxin that is produced during the course of an in vivo infection.
Figure 1. Toxins’ contributions to persistent or acute pathological bacterial conditions. Bacterial genotoxins (BTGXs) and pore-forming toxins (PFTs) can contribute to the modulation of the host’s immune response (IR) during bacterial infection (BI). When BTGXs and PFTs contribute to avoid complete bacterial clearance by the IR and limit immunopathology, the infection can persist for long time, eventually asymptomatically (where chronic carrier status can be associated with pathologies such as cancer). However, when this delicate equilibrium is broken, due to high toxin- and bacterial-induced tissue damage or uncontrolled immune responses, pathological conditions develop.
In this review, we will focus on the immunomodulatory role of two families of bacterial toxins: (a) genotoxins (BTGXs) and (b) pore-forming toxins (PFTs). We have chosen these two effectors, among all the bacterial exotoxins, which may have similar outcomes on the regulation of the host response because they have very different modes of action and target the two innermost and outermost essential structures of a cell, respectively: the DNA and the plasma membrane; the alpha and omega. However, as immunomodulators, they can achieve very similar outcomes, often targeting common node regulators such as transcription factors (e.g., NFκB) and protein kinases, highlighting the relevance of their activities during host–pathogen interactions (Figure 23). For both toxin families, the best-characterised immunomodulatory effect is promotion of proinflammatory responses [2][3][2,4].
Figure 23. Different immunosuppressive effects of BTGXs and PFTs. BTGXs and PFTS can modulate the host’s immune response by several non-mutually exclusive mechanisms: (a) suppression of pro-inflammatory cytokine production/secretion (PFT); (b) re-polarization of the host’s immune response from a pro-inflammatory (Th1 and Th17) to a tolerogenic and tissue repair-prone response (Th2); and (c) toxin expression can reduce the fitness of the bacterium to survive hostile environments such as the blood, favouring tissue colonization and preventing bacteraemia. This can lead to a balance between bacterial infection/colonization (BI) and uncontrolled activation of the immune response (IR).
BTGXs induce DNA damage in target host cells [4][6]. The subsequent cellular response is characterised by secretion of a wide variety of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, and TNFα) within a period of 5 to 72 h post-intoxication in vitro [3][4], possibly via NFκB activation induced by the cellular DNA damage response (DDR) pathway [5][7]. However, there is also a late pro-inflammatory effect (from 4 days post-intoxication onwards), associated with induction of senescence, a permanent quiescent status that prevents growth of DNA-damaged cells. Senescent cells secrete a highly pro-inflammatory secretome, known as senescence-associated secretory phenotype (SASP) [6][7][8,9].
The pro-inflammatory response mediated by PFTs is linked to their capacity to: (i) induce necrosis or pyroptosis, and the subsequent release of damage-associated molecular pattern (DAMPs) into the extracellular environment [8][9][5,10]; (ii) activation of the NLR family pyrin domain-containing 3 (NALP3) inflammasome through diverse mechanisms such as alteration of the ion homeostasis (efflux of potassium K+and influx of calcium Ca2+), or rupture of the phagosome/lysosome compartments, resulting in release of the pro-inflammatory cytokines IL1β and IL-18 [10][11]; (iii) activation of the transcription factor NFκB via induction of Ca2+oscillation at sublethal doses, leading to transcriptional activation and secretion of a broad panel of pro-inflammatory cytokines, such as IL-6 and IL-8 [11][12][12,13].
However, another interesting aspect of the immunomodulatory effects of PFTs and BTGXs is their anti-inflammatory activity, which goes beyond their capacity to induce cell death in general or specifically in cells of the innate or adaptive immune system [3][13][14][4,14,15]. This outcome relies on the manipulation of the immune response, resulting in: (i) suppression of pro-inflammatory cytokines, (ii) re-polarization of the immune response, and (iii) balance between toxin production and bacterial fitness Thus, these effectors can take control and modulate the mechanisms of host tolerance to infection, defined as “reduction of the negative impact of infection on host fitness without affecting the pathogen burden” [15][16] to their own advantage.
2. Bacterial Genotoxins
2.1. General Overview and Mode of Action
Pathogenic bacteria are masters in manipulating the biological processes of infected eukaryotic cells and have the capacity to exploit them for their own advantage. Their main goal is to invade and replicate within the host, possibly in a stealth manner. To achieve the latter, pathogens have developed a variety of weapons during their evolutionary process. Nowadays, one of the most unconventional weapons that bacteria possess is represented by the family of genotoxins.
Bacterial genotoxins are effectors widely diffused among Gram-negative bacteria, which exert their cytotoxic effect by acting as a Type I deoxyribonuclease (DNase I), inducing DNA single-strand breaks (SSBs) and DNA double-strand breaks (DSBs), or as DNA inter-strand cross-linkers. The DNA damaging activity causes cytoplasmic and nuclear distension in targeted cells, eventually leading to cell cycle arrest, apoptosis, or senescence. Three families of genotoxins have been described up to date: the cytolethal distending toxins, the typhoid toxin, and colibactin.
2.1.1. Cytolethal Distending Toxins
The cytolethal distending toxins (CDTs) include a family of bacterial toxins produced by a variety of pathogenic bacteria, such as
Escherichia coli, Aggregatibacter actinomycetemcomitans, Haemophilus ducreyi, Shigella dysenteriae, Campylobacter species, and
Helicobacter species. In order to simplify the specification of CDTs, we use the nomenclature proposed by Cortes-Bratti, where each CDT is referred to by designating the initials of the producing bacterium followed by CDT (e.g.,
Haemophilus ducreyi CDT: HdCDT)
[16][17][17,18].
CDTs are AB2 trimers constituted by the enzymatically active subunit A (CdtB), which is capable of cleaving the DNA of host cells, and B subunits (CdtA and CdtC), which represent the binding components. The entire complex is encoded from a single operon and all three gene products, CdtA, CdtB, and CdtC, are required to confer full activity to the holotoxin.
CdtA and CdtC are lectin-type molecules and represent the binding moiety to the cell surface, an essential mechanism for the subsequent delivery of the active subunit to intracellular compartments. The crystal structure of the CdtB subunit shows the canonical four-layered fold for DNase-I families of enzymes, consisting of a central 12-stranded β-sandwich packed between outer α-helices and loops on each side of the sandwich. In common with DNase I, CdtB possesses two conserved histidine residues (HdCdtB His 160 and His 274), which are critical for toxin activity
[18][19].
The capacity of CDTs to induce cell cycle arrest has been extensively demonstrated in vitro by induction of CDT-mediated fragmentation of purified plasmid
[19][20][20,21] or DNA fragmentation upon intoxication of mammalian cells
[21][22]. As a consequence of DNA damage, cells activate the DNA damage response, leading to cell cycle arrest
[4][22][23][6,23,24].
The mechanisms by which CdtB is internalized and exerts its cytotoxic activity are only partially understood and might differ from CDT to CDT. Several pieces of evidence indicate that membrane cholesterol represents an essential ligand for CDT, which is necessary for the internalization of CdtB and cellular intoxication. This has been proven experimentally by cholesterol depletion from the plasma membrane with methyl-β-cyclodextrin-disrupting lipid rafts, which subsequently abrogated HdCDT,
Actinobacillus actinomycetemcomitans CDT (AaCDT), and
Campilobacter jejuni CDT (CjCDT) binding and intoxication
[24][25][26][25,26,27]. Conversely, the
E. coli CDT (EcCDT) has been reported to bind N-linked fucose-containing complex carbohydrates on cell surfaces
[27][28].
Genes coding for endosomal proteins have been shown to be essential for the cytotoxic activity of CDTs, and thus, it is conceivable that, upon internalization, trafficking via the endosomal compartment is a common feature for many CDTs
[28][29], with the exception of EcCDT, which has been shown to bypass the transit through the late endosome
[29][30].
From the endosomal compartment, AaCDT and HdCDT are retrogradely transported to the Golgi complex and subsequently to the endoplasmic reticulum (ER)
[24][25]. The retrograde transport via the Golgi complex is further supported by Battaglia et al., who demonstrated that AaCdtB translocation is dependent on synaptogyrin-2 and disrupted by Retro-2, an inhibitor of the endosome-Golgi transport
[30][31]. Another key protein that regulates endocytosis and endosomal trafficking is Rab5a, which has been shown to play a role in mediating CdtB-induced inflammatory response and cytotoxicity
[31][32].
The ER to cytosolic translocation of the CdtB subunit may be dependent on the ER-associated degradation (ERAD) pathway
[32][33]. However, the translocation from the ER to the nuclear compartment is still not clear and may differ among several members of the CDT family, as previously suggested
[28][29].
2.1.2. Typhoid Toxin
The genes of the typhoid toxin (TT) have been identified in
S. enterica serovar Typhi, Paratyphi, Schwarzengrund, 9,12:l,v:-, and Bredeney and in
S. enterica subspecies
arizonae and
diarizonae, and
javiana [33][34][35][36][37][34,35,36,37,38]. Different from CDTs, TT possesses an A
2B
5 structure, which includes two enzymatically active subunits and a pentameric ring that is responsible for binding the host cell, allowing the subsequent internalization of the toxin. The active moieties are composed of the CdtB and PltA subunits, which have minimal interaction and are linked by a disulphide bond between Cys214 of PltA and Cys269 of CdtB. Crystallographic studies revealed that PltA exhibits a structure similar to the pertussis-like toxin, with ADP ribosyl transferase activity, whose cellular targets have not been identified yet. CdtB shows a similar structure to the CdtB subunit present in the CDT holotoxin
[34][35]. The binding component is constituted by the pentameric PltB ring, whose five monomers represent the base of the reverse pyramidal structure of the complex
[34][35] and favour the binding to the surface glycoproteins sialoglycans, with an acetyl neuraminic acid as terminal domain, preferentially expressed by human cells
[38][39].
After its receptor-mediated uptake, similar to CDT, TT is retrogradely transported to the ER. In this compartment, the toxin is disassembled by reduction of the disulphide bond between PltA and CdtB, and both components can be individually translocated to the cell cytosol
[39][40].
The typhoid toxin is expressed once the bacterium has been internalized by a host cell, where it is secreted into the lumen of a
Salmonella-containing vacuole, packaged in bacterial outer membrane vesicles (OMVs) that can be secreted via anterograde transport towards the cellular cortex on the microtubule and actin tracks. Upon release into the extracellular environment, the TT-loaded OMVs can be internalized by bystander cells in a dynamin-dependent manner. Induction of DNA damage further requires retrograde transport via the Golgi complex
[40][41].
2.1.3. Colibactin
The genotoxin colibactin is a metabolite produced by the polyketide synthase (
pks) island harboured by extraintestinal pathogenic
E. coli (ExPEC) and other members of
Enterobacteriaceae. The
pks island is present in pathogenic, commensal, and even probiotic bacterial strains
[23][24]. Large-scale whole-genome-based studies have investigated the
pks island phylogeny and suggested the possibility of horizontal acquisition/transmission and exchange between compatible
E. coli subtypes
[41][42].
This metabolite induces double-stranded DNA breaks in eukaryotic cells, causing cell cycle arrest at the G
2-M phase and chromosomal aberrations
[42][43].
Colibactin biosynthesis starts from an assembly line machinery located in the
pks genomic island (54 kb,) which consists of 19 genes comprising non-ribosomal peptide megasynthases (NRPS;
clbH,
clbJ, and
clbN), polyketide megasynthases (PKS;
clbC,
clbI, and
clbO), two hybrid NRPS-PKS (
clbB and
clbK), and nine accessory and tailoring enzymes
[42][43]. A recent study has shown that the production of colibactin’s precursor, precolibactin1489, requires every biosynthetic gene in the colibactin gene cluster, and colibactin is formed through the union of two complex biosynthetic intermediates. This coupling generates a nearly symmetrical structure that contains two electrophilic cyclopropane warheads
[43][44].
Pks-positive
E. coli and a synthetic compound carrying a cyclopropane ring system, similar to that detected in precolibactin candidates, were recently shown to alkylate cellular DNA or purified plasmid DNA, respectively, resulting in extensive DNA fragmentation
[44][45][45,46].
Colibactin-producing bacteria have the capacity to protect themselves from the DNA-damaging action of this metabolite through the expression of ClbS, which deactivates colibactin and confers resistance to its genotoxic effect
[46][47].
The processes of uptake and internalization of colibactin into host cells are still largely unknown, but intoxication requires direct contact between the
pks-positive bacterium and the target cell
[42][47][43,48]. Additionally, the ultimate steps of colibactin’s intracellular trafficking and nuclear access have not been yet identified.
2.1.4. DNA Damage Response
Bacterial genotoxins target the nuclei of host cells, where they inflict their DNA damaging activity. Upon intoxication, eukaryotic cells perceive the damage and initiate the classical DNA damage response (DDR), whereby the cell cycle progression is blocked and repair mechanisms are activated. CDT and TT have the capacity to induce double-strand breaks (DSBs) and single-strand breaks (SSBs), while colibactin induces inter-strand cross-links, which can lead to DSBs. Sensing of damage is mediated by three different kinases, depending on the typology of perturbation: the DNA-dependent protein kinase (DNA-PK) and ataxia telangiectasia mutated (ATM), which mainly detect and respond to DSBs, and ATM-and Rad3 related (ATR), which senses SSBs. In the presence of DNA cross-links, both ATM and ATR are activated
[48][49].
ATM and ATR are recruited to the site of the damaged DNA by the Nbs1–Mre11–Rad50 (MNR) complex or the replication protein A (RPA), respectively. Upon autophosphorylation that activates their kinase activities, both kinases initiate checkpoint responses via the CHK2–p53 axis (ATM) and CHK1–CDC25 axis (ATR), resulting in cell cycle arrest and activation of DNA repair processes. Detailed information summarising the DDR response activated by BTGX can be found elsewhere
[3][4].
The repair machinery consists of three main pathways: (i) non-homologous end joining (NHEJ) repair promoted by DNA-PK, which is carried out by ligation of the two broken DNA ends without requiring a repair template; (ii) homologous repair (HR), in which the resynthesis of the damaged region is accomplished using the undamaged sister chromatid as a template
[49][50][50,51]; and (iii) Fanconi anaemia (FA) pathway to repair DNA inter-strand cross-links. All these systems are activated in response to BTGX intoxication
[51][52][53][52,53,54].
2.2. Biological Functions of BTGXs
In this section, we describe the effects of genotoxin intoxication with a focus on: (i) the outcome when the DNA damage response fails; and (ii) their novel and mostly unexplored effects as anti-inflammatory mediators.
2.2.1. Unresolved DNA Damage Induced by BTGXs
The cell cycle is a series of events that allows cell division and it consists of four phases, defined as G1, S, G2, and M. The G1 and G2 phases are preparatory stages that precede the S phase (synthesis of DNA) and M phase (mitosis), respectively. At the end of each preparatory phase, there is a checkpoint that ensures that all the requirements are completed in order to progress to the next phase: one at the G1/S phases and another at the G2/M phases. Checkpoints can be activated by different events, with DNA damage being a key inducer of these responses, leading to cell cycle arrest and activation of repair machineries. Repaired cells can subsequently resume cell cycle progression. However, if the DNA damage is beyond repair, cells are either eliminated by apoptosis or enter a permanent dormant state known as senescence, activating a tumorigenesis barrier that prevents the proliferation of cells with the high potential to acquire genomic instability and malignant transformation
[7][9].
We briefly discuss here the main features of apoptosis and senescence induced by BTGX.
Apoptosis Induced by Bacterial Genotoxins
The purpose of apoptosis, in this context, is to eliminate defective and damaged cells. Several factors, such as gamma and ultraviolet irradiation, deprivation of growth factors, chemotherapeutic drugs, oncogenes, and viral and bacterial infections, are examples of apoptosis triggering factors
[54][55].
Apoptosis can be initiated by two main mechanisms: the extrinsic pathway that involves death receptor activation (e.g., Fas and TNF receptor 1, TNFR1), leading to activation of caspase 8, and the intrinsic pathway activated by an altered mitochondrial membrane permeability after cellular stress or cellular damage, leading to activation of caspase 9
[55][56]. Several CDTs (AaCDT,
Helicobacter suis, HsCDT, and HdCDT) induce apoptosis in key cells of the immune system such as T lymphocytes (both in primary human purified T cells and the established cell lines Jurkat and MOLT-4
[56][57][58][59][57,58,59,60]) and professional antigen presenting cells, such as macrophages and dendritic cells
[60][61][62][61,62,63]. Two major players in the induction of CDT-induced apoptosis are the tumour suppressor protein TP53 (also known as p53) and members of the caspase family
[55][56].
Exposure of lymphocytes to AaCDT induces activation of caspase 9, which is involved in the initiation of apoptosis via perturbation of the mitochondrial function, and possibly caspase 8 via additional activation of death receptors, such as Fas
[57][58][59][58,59,60]. AaCDT-induced apoptosis can further depend on the blockage of the PI3K signalling cascade and increased levels of the cell cycle inhibitor p21
[63][64][64,65]. Similar patterns of caspase activation have been described for the
Helicobacter hepaticus HhCDT, which upregulated expression of the pro-apoptotic protein Bax with a concomitant decrease in the anti-apoptotic protein Bcl2. Changes in the expression of these apoptotic key players promote the release of mitochondrial cytochrome C and consequently activate caspases 9, 7, and 3 in epithelial cells, the first line of innate defence against pathogens
[65][66].
Interestingly, AaCDT-mediated cell death in the MOLT-4 cell line can be induced by two different pathways: the conventional caspase-dependent apoptosis in the early phase and a caspase-independent apoptosis-like pathway at later stages. Both require the mitochondrial membrane disruption pathway
[59][60], highlighting the complexity of the intoxication-associated regulation of intracellular responses. Whether these described mechanisms are maintained during in vivo scenarios of infection is poorly understood and will require future studies.
BTGXs-Induced Senescence
Upon entry into senescence, cells exhibit characteristic features, such as the formation of promyelocytic leukaemia (PML) nuclear bodies, altered metabolism, and secretion of pro-inflammatory mediators (senescence-associated secretory phenotype, SASP) that remodel the surrounding microenvironment. Even though senescence prevents damaged cells from proliferating, representing a tumorigenesis barrier, long-term senescence is related to chronic inflammation and cancer
[7][9].
Senescence can be exploited by infection agents, as recently reviewed by Humphreys et al.
[66][67]. Induction of senescence, which was transmissible to bystander cells, and secretion of a pro-inflammatory SASP were observed in cells exposed to HdCDT or TT
[67][68][68,69]. The TT-induced senescence enhanced intracellular
Salmonella infection in macrophages and fibroblast-like cells
[68][69], highlighting how bacteria may induce and exploit DNA damage-associated senescence to enhance their invasive capacity.
Induction of BTGX-associated senescence was also proven upon in vitro infection of a broad panel of non-transformed and transformed fibroblasts and epithelial cell lines with
pks+ E. coli [69][70]. The
pks-induced senescent cells presented a SASP, characterised by secretion of pro-inflammatory molecules (IL-6, IL-8, MCP1, MMP3), and the senescent features were transmitted to bystander cells
[69][70]. As proof of the SASP’s tumour-promoting activity, colibactin and HhCDT-induced SASP was also demonstrated to be a key factor in promoting tumour growth in xenograft mouse models
[70][71][72][71,72,73].
2.2.2. Acquisition of Genomic Instability
Genomic instability refers to increased frequencies of genetic alterations and base pair mutations in host cells, and is one of the enabling characteristics of cancer
[73][74].
Induction of genomic instability by long-term exposure to sublethal doses of BTGXs has been extensively studied and demonstrated in experimental models in vitro, highlighting the relevance of the exposure dose and time on the definition of the intoxication outcome.
In vitro exposure to sublethal doses of HdCDT and HhCDT supported malignant cell transformation by increasing the mutation frequency, accumulating chromosomal aberrations and promoting anchorage-independent growth
[74][75]. Furthermore, prolonged infection of Chinese Hamster Ovary (CHO) cells with low doses of
pks+
E. coli revealed a higher percentage of phosphorylated histone H2AX (γH2AX) foci in cell nuclei, as an indicator of DNA damage
[75][76]. Interestingly, unrepaired DSBs resulted in chromatin bridges during the anaphase, lagging chromosomes, and multipolar mitosis, as well as aneuploidy and tetraploidy
[75][76]. Recent studies on colon organoids have shown that short-term infection with
pks+
E. coli promoted the generation of organoids that, due to endogenous Wnt production, which is normally required for cell determination and proliferation
[76][77], grew independently of Wnt supplementation in the culture medium
[77][78], thus resembling aberrant Wnt signalling, which is observed in the majority of colorectal carcinoma (CRC) patients
[78][79][79,80]. Interestingly, the Wnt-independent organoids displayed a higher mutation load and more chromosomal aberrations
[77][78]. This study highlighted the transforming capacities of
pks+
E. coli during early malignant transformation, emphasising the molecular mechanisms that link colibactin-producing bacteria and DNA damage, and supporting their potential carcinogenic properties.
The contribution of genotoxin-producing bacteria on carcinogenesis has been described only in inflammatory or genetic cancer-related in vivo models. Infection of colitis-susceptible
Il10-deficient (
Il10-/-) mice with CDT-deficient
H. hepaticus or
H. cinaedi resulted in less severe typhlocolitis (inflammation of the caecum and adjacent colon) compared with infection with the CDT wild-type isogenic strains
[80][81]. In addition, colonization with the CDT-proficient
H. hepaticus or
H. cinaedi caused intestinal dysplasia and intramucosal carcinoma in the
IL10-/- and
Rag2-/- (T and B cell deficient) models
[81][82][82,83] or liver dysplasia in Swiss Webster mice
[83][84].
Several studies in cancer-prone models have been also performed to assess the carcinogenic potential of
pks+
E. coli. Administration of the pro-carcinogenic agent azoxymethane (AOM, which mimics sporadic CRC) in germ-free
Il10-/- mice showed an enhanced development of adenocarcinomas without affecting inflammation upon monocolonisation with the commensal
pks+
E. coli strain NC101
[84][85]. The pro-inflammatory-prone intestinal environment of
Il10-/- mice further favoured the expansion and maintenance of
Enterobacteriaceae compared with healthy wild-type mice
[85][86].
Interestingly, in this context,
E. coli,
Bacteroides fragilis, Fusobacterium nucleatum,
Parvimonas micra, and
Peptostreptococcus stomatis are enriched in biofilms identified in patients with familial adenomatous polyposis (FAP), a hereditary condition caused by germline mutations of the adenomatous polyposis coli (APC) tumour suppressor gene, frequently mutated in CRC
[86][87][88][87,88,89].
Synergy in colon carcinogenesis between the
pks+
E. coli and
B. fragilis has been demonstrated in two models of sporadic CRC (by administration of AOM) and hereditary CRC (the Apc Min
Δ716/+ mouse model), further corroborating the relevant role of colibactin in colon tumorigenesis
[88][89]. The higher abundance of colibactin-producing bacteria in patient samples from inflammatory bowel disease (IBD) and the most aggressive forms of CRC clinically confirmed the relevance of the in vitro and in vivo findings
[84][89][85,90].
Campylobacter species have also been found to be enriched in CRC samples
[90][91][91,92], and studies on germ-free mice haplo-insufficient for the APC gene (Apc
Min/+) subjected to dextran sodium sulphate (DSS) reported the development of large tumours in the distal colon of mice infected with a
C. jejuni strain carrying a functional CdtB subunit
[92][93].
2.2.3. Immunomodulatory Properties of BTGX
Considering that DNA damage is regarded as a danger for the cell homeostasis, it is expected that infection with genotoxin-producing bacteria will promote a pro-inflammatory response. Indeed, several studies have demonstrated the pro-inflammatory effects of BTGX, such as Hh-CDT, Hp-CDT, Aa-CDT, CjCDT, and colibactin-producing
E. coli in vitro
[31][93][94][95][96][97][98][99][32,94,95,96,97,98,99,100] and in vivo
[81][82][83][100][101][102][103][104][105][82,83,84,101,102,103,104,105,106].
The DNA-damage induced pro-inflammatory response can be activated by four pathways, which are not mutually exclusive: (i) NFκB activation
[103][104], possibly via the ATM kinase pathway
[5][7]; (ii) mitogen-activated protein kinase (MAPK), p38, and signal transducer and activator of transcription (STAT3) phosphorylation
[82][106][83,107], which lead to the production of pro-inflammatory cytokines, IL-1β, IL-6, IL-8, and TNF-α
[107][108][109][108,109,110]; (iii) formation of by-products of mitotic progression (micronuclei), which activate cGAS-STING signalling to further secrete Type I interferons
[110][111][112][113][111,112,113,114]; and (iv) secretion of SASP, characterised by IL-1α, IL-6, IL-8, MMP-1, and MMP-3
[7][67][68][69][9,68,69,70]. Together, these secreted pro-inflammatory cytokines further activate pro-inflammatory and efficient anti-bacterial T helper (Th)1 and Th17 immune responses
[93][114][115][116][94,115,116,117]. The underlying activation mechanisms of these pathways have been recently reviewed by Martin and Frisan
[3][4], and will not be described here in detail.
Here, we will focus on describing the anti-inflammatory and immunosuppressive roles of BTGXs, which have not been as widely reviewed.
It has been shown that both AaCDT and HdCDT block the proliferation of T lymphocytes by inducing G2/M cell cycle arrest and apoptosis, suggesting an immunosuppressive role of these toxins
[56][117][57,118]. Furthermore,
pks+ extraintestinal
E. coli (ExPEC) induced DNA damage, cell cycle arrest in the G2/M phase, and cell death in T lymphocytes in vitro
[118][119]. Induction of cell death and cell cycle arrest in lymphocytes may significantly affect the course of infection, favouring the bacterial dissemination, multiplication, and persistence of BTGX-carrying pathogens
[119][120]. Thus, the immunosuppressive effect of colibactin may explain the increased mortality induced by infection with a
pks+
E. coli compared with an isogenic colibactin-deficient strain (Δ
clbA) strain in a mouse model of sepsis
[118][119].
The anti-inflammatory effect of BTGXs has been also shown for the
pks+ probiotic
E. coli strain Nissle 1917, where deletion of the
cblA gene abolished the genotoxin effect in vitro, as well as the anti-inflammatory properties of this strain in two independent models of colitis
[120][121]. However, considering the complexity of the
pks island, to date, it has not been possible to define unequivocally whether the immunosuppressive effect of the Nissle strain is directly mediated by the DNA-damaging activity of colibactin. Therefore, additional experiments including mouse models deficient in a functional DDR, such as ATM-deficient mice, could help to address this important and interesting question, thereby shedding light on the underlying mechanisms involved.
Infection studies performed with a TT-expressing
S. enterica have shown the suppression of gastroenteritis characterised by a decrease in the recruitment of CD45
+ (leukocytes), CD3
+ (T lymphocytes), and F4/80
+ (macrophages) cells
[121][122][123][122,123,124]. The presence of a functional TT promoted the induction of a Th2 tissue-protective immune response characterised by the presence of anti-inflammatory CD206
+ macrophages and T regulatory lymphocytes, and enhanced expression of mRNA for Th2 cytokines (
Il10,
Il4,
Il13,
Il5)
[123][124]. These anti-inflammatory effects were partially ATM-dependent and occurred in spite of the significant induction of DNA fragmentation in the colon mucosa and the substantial presence of senescent cells, supporting the idea that, under these conditions, DNA damage and senescence are uncoupled from inflammation.
Interestingly, a functional TT did not prevent inflammation in the liver and spleen
[121][122], and the anti-inflammatory effects were lost when infection occurred in mice suffering from acute DSS-induced colitis
[123][124]. Therefore, the described effects of a functional TT were context- (healthy vs. colitis) and tissue- (intestine vs. liver and spleen) dependent.