 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Driss LAGHLAM | + 1621 word(s) | 1621 | 2021-07-22 03:47:44 | | | |
| 2 | Enzi Gong | Meta information modification | 1621 | 2021-08-05 02:40:26 | | |
Video Upload Options
The renin–angiotensin system (RAS) has long been considered the pinnacle of homeostasis in cardiovascular physiology. Its main function involves regulation of blood pressure, via direct and indirect means, through electrolyte balance, and trophic and vasomotor functions. While historically associated with these basic functions, the role of inflammation in cardiovascular diseases has been more and more described, and a critical role of RAS in inflammation regulation has been suggested.
1. Overview
The renin–angiotensin system (RAS) has long been described in the field of cardiovascular physiology as the main player in blood pressure homeostasis. However, other effects have since been described, and include proliferation, fibrosis, and inflammation. To illustrate the immunomodulatory properties of the RAS, we chose three distinct fields in which RAS may play a critical role and be the subject of specific treatments. In oncology, RAS hyperactivation has been associated with tumor migration, survival, cell proliferation, and angiogenesis; preliminary data showed promise of the benefit of RAS blockers in patients treated for certain types of cancer. In intensive care medicine, vasoplegic shock has been associated with severe macro- and microcirculatory imbalance. A relative insufficiency in angiotensin II (AngII) was associated to lethal outcomes and synthetic AngII has been suggested as a specific treatment in these cases. Finally, in solid organ transplantation, both AngI and AngII have been associated with increased rejection events, with a regional specificity in the RAS activity. These elements emphasize the complexity of the direct and indirect interactions of RAS with immunomodulatory pathways and warrant further research in the field.
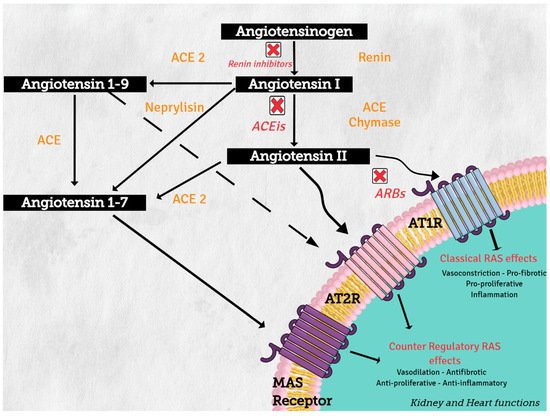
2. Renin–Angiotensin System
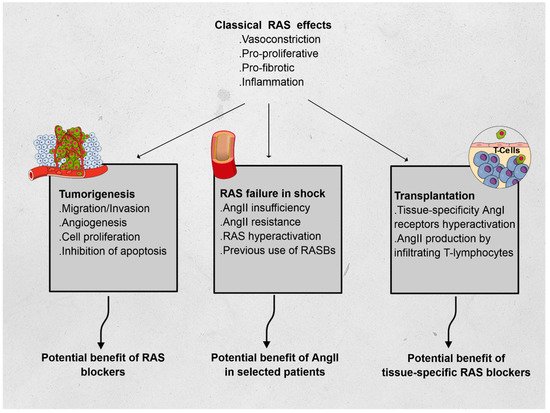
3. Pathways Related to RAS
4. RAS, Inflammation, Diabetes, and Metabolic Syndrome
References
- de Gasparo, M.; Catt, K.J.; Inagami, T.; Wright, J.W.; Unger, T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol. Rev. 2000, 52, 415–472.
- Paz Ocaranza, M.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129.
- Passos-Silva, D.G.; Verano-Braga, T.; Santos, R.A. Angiotensin-(1–7): Beyond the cardio-renal actions. Clin. Sci. (Lond.) 2013, 124, 443–456.
- Ocaranza, M.P.; Moya, J.; Barrientos, V.; Alzamora, R.; Hevia, D.; Morales, C.; Pinto, M.; Escudero, N.; García, L.; Novoa, U.; et al. Angiotensin-(1–9) reverses experimental hypertension and cardiovascular damage by inhibition of the angiotensin converting enzyme/Ang II axis. J. Hypertens. 2014, 32, 771–783.
- Drummond, G.R.; Vinh, A.; Guzik, T.J.; Sobey, C.G. Immune mechanisms of hypertension. Nat. Rev. Immunol. 2019, 19, 517–532.
- Mikolajczyk, T.P.; Guzik, T.J. Adaptive Immunity in Hypertension. Curr. Hypertens. Rep. 2019, 21, 68.
- Xiao, L.; do Carmo, L.S.; Foss, J.D.; Chen, W.; Harrison, D.G. Sympathetic Enhancement of Memory T-Cell Homing and Hypertension Sensitization. Circ. Res. 2020, 126, 708–721.
- Marvar, P.J.; Harrison, D.G. Stress-dependent hypertension and the role of T lymphocytes. Exp. Physiol. 2012, 97, 1161–1167.
- Zhao, J.V.; Schooling, C.M.; Leung, G.M. Using genetics to understand the role of antihypertensive drugs modulating angiotensin-converting enzyme in immune function and inflammation. Br. J. Clin. Pharmacol. 2021, 87, 1839–1846.
- Qin, X.Y.; Zhang, Y.L.; Chi, Y.F.; Yan, B.; Zeng, X.J.; Li, H.H.; Liu, Y. Angiotensin II Regulates Th1 T Cell Differentiation Through Angiotensin II Type 1 Receptor-PKA-Mediated Activation of Proteasome. Cell. Physiol. Biochem. 2018, 45, 1366–1376.
- Hunyady, L.; Catt, K.J. Pleiotropic AT1 receptor signaling pathways mediating physiological and pathogenic actions of angiotensin II. Mol. Endocrinol. 2006, 20, 953–970.
- Balakumar, P.; Jagadeesh, G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell. Signal. 2014, 26, 2147–2160.
- Mehta, P.K.; Griendling, K.K. Angiotensin II cell signaling: Physiological and pathological effects in the cardiovascular system. Am. J. Physiol. Cell Physiol. 2007, 292, C82–C97.
- Anavekar, N.S.; Solomon, S.D. Angiotensin II receptor blockade and ventricular remodelling. J. Renin Angiotensin Aldosterone Syst. 2005, 6, 43–48.
- Suzuki, K.; Han, G.D.; Miyauchi, N.; Hashimoto, T.; Nakatsue, T.; Fujioka, Y.; Koike, H.; Shimizu, F.; Kawachi, H. Angiotensin II type 1 and type 2 receptors play opposite roles in regulating the barrier function of kidney glomerular capillary wall. Am. J. Pathol. 2007, 170, 1841–1853.
- Niu, M.J.; Yang, J.K.; Lin, S.S.; Ji, X.J.; Guo, L.M. Loss of angiotensin-converting enzyme 2 leads to impaired glucose homeostasis in mice. Endocrine 2008, 34, 56–61.
- Patel, V.B.; Mori, J.; McLean, B.A.; Basu, R.; Das, S.K.; Ramprasath, T.; Parajuli, N.; Penninger, J.M.; Grant, M.B.; Lopaschuk, G.D.; et al. ACE2 Deficiency Worsens Epicardial Adipose Tissue Inflammation and Cardiac Dysfunction in Response to Diet-Induced Obesity. Diabetes 2016, 65, 85–95.
- Coppo, M.; Bandinelli, M.; Chiostri, M.; Poggesi, L.; Boddi, M. T-Lymphocyte-Based Renin Angiotensin System in Obesity. Am. J. Med. Sci. 2019, 358, 51–58.
- Bindom, S.M.; Hans, C.P.; Xia, H.; Boulares, A.H.; Lazartigues, E. Angiotensin I-converting enzyme type 2 (ACE2) gene therapy improves glycemic control in diabetic mice. Diabetes 2010, 59, 2540–2548.
- Osterreicher, C.H.; Taura, K.; De Minicis, S.; Seki, E.; Penz-Osterreicher, M.; Kodama, Y.; Kluwe, J.; Schuster, M.; Oudit, G.Y.; Penninger, J.M.; et al. Angiotensin-converting-enzyme 2 inhibits liver fibrosis in mice. Hepatology 2009, 50, 929–938.
- Cao, X.; Yang, F.; Shi, T.; Yuan, M.; Xin, Z.; Xie, R.; Li, S.; Li, H.; Yang, J.K. Angiotensin-converting enzyme 2/angiotensin-(1–7)/Mas axis activates Akt signaling to ameliorate hepatic steatosis. Sci. Rep. 2016, 6, 21592.
- Munoz, M.C.; Giani, J.F.; Dominici, F.P. Angiotensin-(1–7) stimulates the phosphorylation of Akt in rat extracardiac tissues in vivo via receptor Mas. Regul. Pept. 2010, 161, 1–7.
- Feltenberger, J.D.; Andrade, J.M.; Paraiso, A.; Barros, L.O.; Filho, A.B.; Sinisterra, R.D.; Sousa, F.B.; Guimaraes, A.L.; de Paula, A.M.; Campagnole-Santos, M.J.; et al. Oral formulation of angiotensin-(1–7) improves lipid metabolism and prevents high-fat diet-induced hepatic steatosis and inflammation in mice. Hypertension 2013, 62, 324–330.
- Santos, S.H.; Andrade, J.M.; Fernandes, L.R.; Sinisterra, R.D.; Sousa, F.B.; Feltenberger, J.D.; Alvarez-Leite, J.I.; Santos, R.A. Oral Angiotensin-(1–7) prevented obesity and hepatic inflammation by inhibition of resistin/TLR4/MAPK/NF-kappaB in rats fed with high-fat diet. Peptides 2013, 46, 47–52.
- Santos, S.H.; Fernandes, L.R.; Pereira, C.S.; Guimaraes, A.L.; de Paula, A.M.; Campagnole-Santos, M.J.; Alvarez-Leite, J.I.; Bader, M.; Santos, R.A. Increased circulating angiotensin-(1–7) protects white adipose tissue against development of a proinflammatory state stimulated by a high-fat diet. Regul. Pept. 2012, 178, 64–70.
- Liu, C.; Lv, X.H.; Li, H.X.; Cao, X.; Zhang, F.; Wang, L.; Yu, M.; Yang, J.K. Angiotensin-(1–7) suppresses oxidative stress and improves glucose uptake via Mas receptor in adipocytes. Acta Diabetol. 2012, 49, 291–299.
- Santos, S.H.; Braga, J.F.; Mario, E.G.; Porto, L.C.; Rodrigues-Machado Mda, G.; Murari, A.; Botion, L.M.; Alenina, N.; Bader, M.; Santos, R.A. Improved lipid and glucose metabolism in transgenic rats with increased circulating angiotensin-(1–7). Arterioscler. Thromb. Vasc. Biol. 2010, 30, 953–961.
- Santos, S.H.; Fernandes, L.R.; Mario, E.G.; Ferreira, A.V.; Porto, L.C.; Alvarez-Leite, J.I.; Botion, L.M.; Bader, M.; Alenina, N.; Santos, R.A. Mas deficiency in FVB/N mice produces marked changes in lipid and glycemic metabolism. Diabetes 2008, 57, 340–347.
- Saiki, A.; Ohira, M.; Endo, K.; Koide, N.; Oyama, T.; Murano, T.; Watanabe, H.; Miyashita, Y.; Shirai, K. Circulating angiotensin II is associated with body fat accumulation and insulin resistance in obese subjects with type 2 diabetes mellitus. Metab. Clin. Exp. 2009, 58, 708–713.
- Wei, Y.; Clark, S.E.; Morris, E.M.; Thyfault, J.P.; Uptergrove, G.M.; Whaley-Connell, A.T.; Ferrario, C.M.; Sowers, J.R.; Ibdah, J.A. Angiotensin II-induced non-alcoholic fatty liver disease is mediated by oxidative stress in transgenic TG(mRen2)27(Ren2) rats. J. Hepatol. 2008, 49, 417–428.
- Wei, Y.; Sowers, J.R.; Nistala, R.; Gong, H.; Uptergrove, G.M.; Clark, S.E.; Morris, E.M.; Szary, N.; Manrique, C.; Stump, C.S. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells. J. Biol. Chem. 2006, 281, 35137–35146.
- Chou, C.L.; Lin, H.; Chen, J.S.; Fang, T.C. Renin inhibition improves metabolic syndrome, and reduces angiotensin II levels and oxidative stress in visceral fat tissues in fructose-fed rats. PLoS ONE 2017, 12, e0180712.

