The renin–angiotensin system (RAS) has long been considered the pinnacle of homeostasis in cardiovascular physiology. Its main function involves regulation of blood pressure, via direct and indirect means, through electrolyte balance, and trophic and vasomotor functions. While historically associated with these basic functions, the role of inflammation in cardiovascular diseases has been more and more described, and a critical role of RAS in inflammation regulation has been suggested.
1. Overview
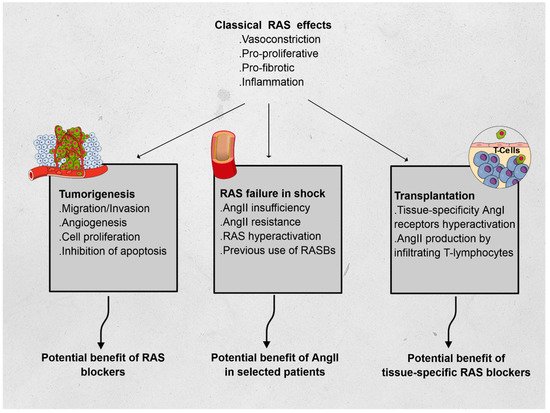
The renin–angiotensin system (RAS) has long been described in the field of cardiovascular physiology as the main player in blood pressure homeostasis. However, other effects have since been described, and include proliferation, fibrosis, and inflammation. To illustrate the immunomodulatory properties of the RAS, we chose three distinct fields in which RAS may play a critical role and be the subject of specific treatments. In oncology, RAS hyperactivation has been associated with tumor migration, survival, cell proliferation, and angiogenesis; preliminary data showed promise of the benefit of RAS blockers in patients treated for certain types of cancer. In intensive care medicine, vasoplegic shock has been associated with severe macro- and microcirculatory imbalance. A relative insufficiency in angiotensin II (AngII) was associated to lethal outcomes and synthetic AngII has been suggested as a specific treatment in these cases. Finally, in solid organ transplantation, both AngI and AngII have been associated with increased rejection events, with a regional specificity in the RAS activity. These elements emphasize the complexity of the direct and indirect interactions of RAS with immunomodulatory pathways and warrant further research in the field.
2. Renin–Angiotensin System
The renin–angiotensin system (RAS) has long been considered the pinnacle of homeostasis in cardiovascular physiology. Its main function involves regulation of blood pressure, via direct and indirect means, through electrolyte balance, and trophic and vasomotor functions. While historically associated with these basic functions, the role of inflammation in cardiovascular diseases has been more and more described, and a critical role of RAS in inflammation regulation has been suggested.
Using three different fields of application, we hereafter describe non-classical RAS functions, relying on the immunomodulatory features of RAS: oncology, intensive care, and transplantation (see Figure 1).
Figure 1. Effects of the renin–angiotensin system (RAS) and its features in oncology, vasoplegic shock, and transplantation. Abbreviations: AngI: angiotensin I, AngII: angiotensin II.
3. Pathways Related to RAS
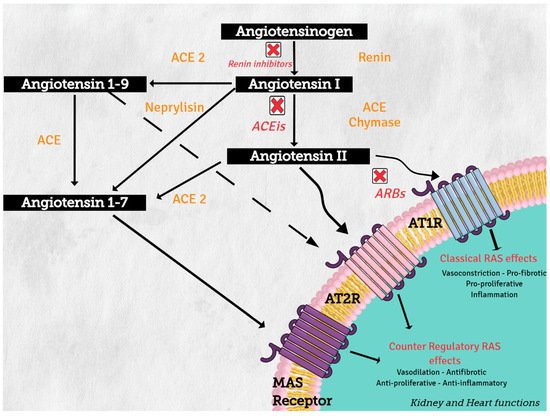
Angiotensinogen (synthesized in the liver) is the only precursor protein of RAS and the source of all angiotensin peptides. Angiotensinogen is primarily cleaved in blood circulation by renin (protease synthesized in the kidney), to angiotensin I (AngI), which in turn is processed by angiotensin converting enzyme (ACE) (secreted in lung) to produce the active octapeptide, angiotensin II (AngII). AngII actions are primary mediated through the binding of its two receptors, type 1 (AT1R) and type 2 (AT2R), which belong to the G protein-coupled receptor superfamily. The important cardiovascular actions of AngII, including regulation of arterial blood pressure with short-term vasoconstriction, aldosterone release, and water and salt balance, are predominantly mediated by AT1R in target tissues, such as blood vessels, kidney, brain, and heart
[1].
On the other hand, increasing evidence supports the concept of a counter regulatory renin–angiotensin system with opposite effects in cardiovascular physiology and disease
[2]. Angiotensin converting enzyme 1 (ACE2) and neprilysin (NEP) are part of this counter regulatory system. ACE2 and NEP can cleave AngII to produce angiotensin 1–7 (Ang 1–7), and ACE2 cleaves AngI to generate angiotensin 1–9 (Ang 1–9) (see
Figure 2). These two peptides mediate the antagonistic effects of AngII. Angiotensin 1–7 binds to the proto-oncogen Mas receptor, which leads to vasodilation, antihypertensive, and antifibrosis effects
[3], while Ang 1–9 can activate AT2R to trigger natriuresis and NO production, thus mediating vasodilatory effects and reducing blood pressure
[4].
Figure 2. Renin–angiotensin system: classical and counter regulatory pathways. Abbreviations: ACE: angiotensin converting enzyme; ACE 2: angiotensin-converting enzyme type 2, ACEIs: angiotensin-converting enzyme inhibitors; ARBs: angiotensin-2 receptor 1 blockers, AT1R: angiotensin II receptor type1, AT2R: angiotensin II receptor type 2.
Over the past decade, growing evidence has shown that an altered immune system is likely to be a key contributor to the development of hypertension. Consistent evidence demonstrates that various immune cell subsets infiltrate blood vessels, kidneys, heart, and/or the autonomic nervous system during hypertension, and that the targeted depletion of specific immune cell subsets or inhibition of their functions protects against hypertension in animal models. However, no definitive mechanistic explanation has been established for how immune cells influence organ functions to promote increases in blood pressure
[5]. Pro-inflammatory T cell-derived cytokines, such as IFN-γ and TNF-α (from CD8+ and CD4 + Th1) and IL-17A (from the γδ-T cell and CD4 + Th17), were found to exacerbate hypertensive responses mediating both endothelial dysfunction and cardiac, renal, and neurodegenerative injury. B cell activation and production of autoantibodies (anti-Hsp70, anti-Hsp65, anti-Hsp60, anti-AT1R, anti-α1AR, and anti-β1AR) were also associated with hypertension, while hypertensive responses can be inhibited by T regulatory lymphocytes (Tregs) and the anti-inflammatory IL-10
[6]. Other elements have been associated with a modulation of response of lymphocytes T to AngII, most lately sympathetic tone
[7], as suggested by stress-related hypertension
[8].
The role of antihypertensive drugs in modulating angiotensin-converting enzyme in immune function and inflammation could be genetically related. A recent study gave indications of the beneficial effects of ACEIs on immune function and inflammation, using a Mendelian randomization in the largest available genome-wide association studies, genetically predicted the effects of ACEI’s (single nucleotide polymorphisms rs4968783 and rs4291 in ACE) increased lymphocyte percentage (0.78, 95% confidence interval (CI) 0.35, 1.22) and decreased neutrophil percentage (−0.64, 95% CI −1.09, −0.20), and possibly lowered TNF-α (−4.92, 95% CI −8.50, −1.33). This effect of ACEIs is not due to, or at least is not totally due to ACE 2 expression, because, in contrast ARBs, which also affect ACE2 expression, they were not found in this study to affect lymphocyte, neutrophil, or TNF-α
[9].
Qin et al. demonstrated that AngII induces the expression of proteasome protein subunits and predominantly stimulates Th1 differentiation. This effect of AngII in proteasome activities was associated with the activation of the AT1R/PKA signaling pathway, which in turn promotes the degradation of MAKP-1 (mitogen-activated protein kinase) and IκBα and the activation of the STAT1 and NF-κB pathways, which eventually lead to Th1 cell differentiation. In addition, inhibition of proteasome activation or AT1R markedly attenuated these effects
[10].
The activated AT1R coupled with a classical Gq/11 protein can also activate multiple intracellular signal transduction pathways in cardiovascular, renal, and immune cells; with several tyrosine kinases, including receptor tyrosine kinases (epidermal growth factor receptor (EGFR), PDGF, and insulin receptor) and nonreceptor tyrosine kinases (c-Src family kinases, Ca2-dependent proline-rich tyrosine kinase 2 (Pyk2), focal adhesion kinase (FAK), and Janus kinases (JAK)). AT1R also activates serine/threonine kinases, such as PKC and MAPK (extracellular regulated kinases (ERK) 1/2, p38MAPK and Jun N-terminal kinase). Transcription of nuclear factor κB (NFκB) participates in the Raf pathway and generates ROS via NAD(P)H oxidase activation (in cardiovascular cells, leukocytes, and monocytes)
[11][12][13][11,12,13]. This link with tyrosine kinase-related pathways and their downstream effectors explains the well-established role of RAS in the growth and remodeling of the vasculature, kidney, and heart, which occurs during metabolic disturbances and cardiovascular disease
[13][14][13,14]. Although AT1R mediates most of the known vasoactive effects of AngII, AT2R via the activation of phosphatases, GMPc, and phospholipase-A2, and mainly by antagonizing AT1Rs, contribute to the regulation of blood pressure and renal function and decrease cell proliferation in the kidney glomerular capillary wall while increasing apoptosis and cell differentiation
[15].
4. RAS, Inflammation, Diabetes, and Metabolic Syndrome
It must be emphasized that the association between RAS and inflammation has also been observed in the context of metabolic syndrome, including diabetes, mostly through ACE2 activity. Indeed, ACE2 is expressed in liver, skeletal muscles, and adipose tissue, and any disturbance to ACE2/Ang-(1–7) activity could lead to a glucose homeostasis disorder. Moreover, loss of the ACE2 gene in mice leads to hepatic fibrosis and impaired glucose homeostasis through an elevated hepatic reactive oxygen species level, an increased oxidative stress, and inflammation in the liver, leading to an impairment in insulin signaling
[16]. In adipose tissue, ACE2 deficiency worsens inflammation in response to diet-induced obesity in mice
[17]. Incidentally, RAS activity in T-cells was also found to be disturbed in obese patients, in whom AngII stimulation was observed
[18]. Conversely, overexpression of ACE2 or Ang-(1–7) administration improves these metabolic disorders, i.e., glycemic control and insulin sensitivity
[19][20][21][19,20,21]. Indeed, mechanistically, Ang-(1–7) rescues the insulin signaling pathway by stimulating PKB phosphorylation, a main mediator of the insulin signaling pathway, which will then activate the downstream glycogen synthase kinase-3β in the liver and skeletal muscles, resulting in a decrease in glycemia through glycogen storage
[22] in several murine models of diet-induced insulin resistance, such as high-fat diet fed mice or in fructose-fed rats
[23][24][23,24]. In adipose tissue, activation of ACE2/Ang-(1–7) prevents inflammation and oxidative stress induced by a high-fat diet and increases glucose uptake and adiponectin level
[25][26][27][25,26,27], while its disturbance results in a lower insulin-dependent glucose uptake and adiponectin secretion
[28].
In addition to the effects of the inhibition of the above-mentioned alternate effects of RAS, in the context of metabolic diseases, such as obesity, T2DM, or nonalcoholic fatty liver disease, plasmatic Ang II is positively correlated with body weight and is associated with insulin resistance, suggesting that ACE/Ang II activity is upregulated in those metabolic disorders
[29]. In addition, on a tissue scale, Ang II was associated with increased insulin resistance through oxidative stress, leading to hepatic fibrosis and cirrhosis, provoking an impairment of insulin signaling
[30]. In skeletal muscles, Ang II also induces a decreased glucose uptake and impairs insulin sensitivity
[31], while in adipose tissue, it inhibits adiponectin secretion and insulin signaling through an increased oxidative stress
[32]. These elements emphasize the pro-diabetogenic effects of classical RAS effects, in parallel to those of inflammation.