 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Piero Portincasa | + 2348 word(s) | 2348 | 2021-07-26 08:59:39 | | | |
| 2 | Lily Guo | Meta information modification | 2348 | 2021-08-09 06:21:08 | | | | |
| 3 | Conner Chen | Meta information modification | 2348 | 2021-10-12 10:58:08 | | |
Video Upload Options
The liver plays a key role in systemic metabolic processes, which include detoxification, synthesis, storage, and export of carbohydrates, lipids, and proteins. The raising trends of obesity and metabolic disorders worldwide is often associated with the nonalcoholic fatty liver disease (NAFLD), which has become the most frequent type of chronic liver disorder with risk of progression to cirrhosis and hepatocellular carcinoma. Liver mitochondria play a key role in degrading the pathways of carbohydrates, proteins, lipids, and xenobiotics, and to provide energy for the body cells. The morphological and functional integrity of mitochondria guarantee the proper functioning of β-oxidation of free fatty acids and of the tricarboxylic acid cycle.
1. Introduction
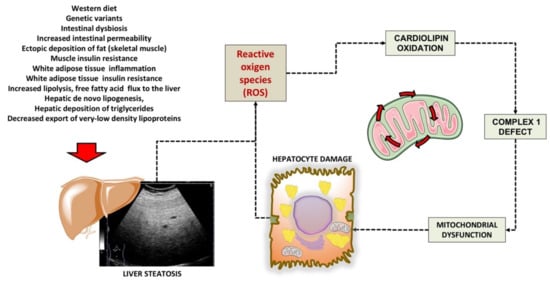
The liver plays a key role in lipid homeostasis, with steps including the synthesis, oxidation, and transport of free fatty acids (FFA), triglycerides (TG), cholesterol, and bile acids (BA). Chronic liver diseases encompass a spectrum of conditions ranging from metabolic to viral, alcohol-related diseases, drug-related diseases, autoimmune diseases, and tumours. The hepatocyte can be damaged by various hits and intracellular organelles can be part of the dysfunctional cell, with changes including microsomal hypertrophy, mitochondrial damage by free fatty acids overload and insufficient β-oxidation and activation of peroxisomal metabolism.
Growing evidence points to dysfunctional mitochondria as key contributors in the pathogenesis of the chronic metabolic conditions (i.e., obesity, metabolic syndrome and type 2 diabetes mellitus) frequently linked to liver disease. These processes act through pathways leading to oxidative stress, chronic inflammation, and insulin resistance. Thus, diagnostic techniques able to early detect and monitor mitochondrial dysfunction have great relevance in terms of possible primary/secondary prevention measures and of therapies specifically targeting liver mitochondria [1,2,3].
2. Mitochondrial Dysfunction in the Liver
References
- Caldwell, S.H.; Swerdlow, R.H.; Khan, E.M.; Iezzoni, J.C.; Hespenheide, E.E.; Parks, J.K.; Parker, W.D., Jr. Mitochondrial abnormalities in non-alcoholic steatohepatitis. J. Hepatol. 1999, 31, 430–434.
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468.
- Simoes, I.C.M.; Karkucinska-Wieckowska, A.; Janikiewicz, J.; Szymanska, S.; Pronicki, M.; Dobrzyn, P.; Dabrowski, M.; Dobrzyn, A.; Oliveira, P.J.; Zischka, H.; et al. Western Diet Causes Obesity-Induced Nonalcoholic Fatty Liver Disease Development by Differentially Compromising the Autophagic Response. Antioxidants 2020, 9, 995.
- Longo, M.; Meroni, M.; Paolini, E.; Macchi, C.; Dongiovanni, P. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): New perspectives for a fairy-tale ending? Metab. Clin. Exp. 2021, 117, 154708.
- Ajaz, S.; McPhail, M.J.; Gnudi, L.; Trovato, F.M.; Mujib, S.; Napoli, S.; Carey, I.; Agarwal, K. Mitochondrial dysfunction as a mechanistic biomarker in patients with non-alcoholic fatty liver disease (NAFLD). Mitochondrion 2021, 57, 119–130.
- Shannon, C.E.; Ragavan, M.; Palavicini, J.P.; Fourcaudot, M.; Bakewell, T.M.; Valdez, I.A.; Ayala, I.; Jin, E.S.; Madesh, M.; Han, X.; et al. Insulin resistance is mechanistically linked to hepatic mitochondrial remodeling in non-alcoholic fatty liver disease. Mol. Metab. 2021, 45, 101154.
- Li, Y.; Wu, J.; Yang, M.; Wei, L.; Wu, H.; Wang, Q.; Shi, H. Physiological evidence of mitochondrial permeability transition pore opening caused by lipid deposition leading to hepatic steatosis in db/db mice. Free Radic. Biol. Med. 2021, 162, 523–532.
- Yan, C.; Duanmu, X.; Zeng, L.; Liu, B.; Song, Z. Mitochondrial DNA: Distribution, Mutations, and Elimination. Cells 2019, 8, 379.
- Wei, Y.; Rector, R.S.; Thyfault, J.P.; Ibdah, J.A. Nonalcoholic fatty liver disease and mitochondrial dysfunction. World J. Gastroenterol. WJG 2008, 14, 193–199.
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864.
- Pan, J.; Ou, Z.; Cai, C.; Li, P.; Gong, J.; Ruan, X.Z.; He, K. Fatty acid activates NLRP3 inflammasomes in mouse Kupffer cells through mitochondrial DNA release. Cell Immunol. 2018, 332, 111–120.
- Pirola, C.J.; Garaycoechea, M.; Flichman, D.; Castano, G.O.; Sookoian, S. Liver mitochondrial DNA damage and genetic variability of Cytochrome b—A key component of the respirasome drive the severity of fatty liver disease. J. Intern. Med. 2021, 289, 84–96.
- Malik, A.N.; Czajka, A. Is mitochondrial DNA content a potential biomarker of mitochondrial dysfunction? Mitochondrion 2013, 13, 481–492.
- Begriche, K.; Massart, J.; Robin, M.A.; Bonnet, F.; Fromenty, B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology 2013, 58, 1497–1507.
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxidative Med. Cell. Longev. 2018, 2018, 9547613.
- Seki, S.; Kitada, T.; Yamada, T.; Sakaguchi, H.; Nakatani, K.; Wakasa, K. In situ detection of lipid peroxidation and oxidative DNA damage in non-alcoholic fatty liver diseases. J. Hepatol. 2002, 37, 56–62.
- Weltman, M.D.; Farrell, G.C.; Liddle, C. Increased hepatocyte CYP2E1 expression in a rat nutritional model of hepatic steatosis with inflammation. Gastroenterology 1996, 111, 1645–1653.
- Chalasani, N.; Gorski, J.C.; Asghar, M.S.; Asghar, A.; Foresman, B.; Hall, S.D.; Crabb, D.W. Hepatic cytochrome P450 2E1 activity in nondiabetic patients with nonalcoholic steatohepatitis. Hepatology 2003, 37, 544–550.
- Aubert, J.; Begriche, K.; Knockaert, L.; Robin, M.A.; Fromenty, B. Increased expression of cytochrome P450 2E1 in nonalcoholic fatty liver disease: Mechanisms and pathophysiological role. Clin. Res. Hepatol. Gastroenterol. 2011, 35, 630–637.
- Caballero, F.; Fernandez, A.; Matias, N.; Martinez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernandez-Checa, J.C.; Garcia-Ruiz, C. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J. Biol. Chem. 2010, 285, 18528–18536.
- Gao, D.; Wei, C.; Chen, L.; Huang, J.; Yang, S.; Diehl, A.M. Oxidative DNA damage and DNA repair enzyme expression are inversely related in murine models of fatty liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1070–G1077.
- Tesse, A.; Grossini, E.; Tamma, G.; Brenner, C.; Portincasa, P.; Marinelli, R.A.; Calamita, G. Aquaporins as targets of dietary bioactive phytocompounds. Front. Mol. Biosci. 2018, 5, 30.
- Calamita, G.; Portincasa, P. The power of science diplomacy, a lesson from the Nobel laureate Peter Agre. Eur. J. Clin. Investig. 2016, 46, 491–493.
- Portincasa, P.; Palasciano, G.; Svelto, M.; Calamita, G. Aquaporins in the hepatobiliary tract. Which, where and what they do in health and disease. Eur. J. Clin. Investig. 2008, 38, 1–10.
- Ferri, D.; Mazzone, A.; Liquori, G.E.; Cassano, G.; Svelto, M.; Calamita, G. Ontogeny, distribution, and possible functional implications of an unusual aquaporin, AQP8, in mouse liver. Hepatology 2003, 38, 947–957.
- Marchissio, M.J.; Frances, D.E.; Carnovale, C.E.; Marinelli, R.A. Mitochondrial aquaporin-8 knockdown in human hepatoma HepG2 cells causes ROS-induced mitochondrial depolarization and loss of viability. Toxicol. Appl. Pharmacol. 2012, 264, 246–254.
- Busanello, E.N.B.; Figueira, T.R.; Marques, A.C.; Navarro, C.D.C.; Oliveira, H.C.F.; Vercesi, A.E. Facilitation of Ca(2+) -induced opening of the mitochondrial permeability transition pore either by nicotinamide nucleotide transhydrogenase deficiency or statins treatment. Cell Biol. Int. 2018, 42, 742–746.
- Slater, T.F. Free radical mechanisms in tissue injury. Cell Funct. Dis. 1988, 222, 1–15.
- Petrosillo, G.; Portincasa, P.; Grattagliano, I.; Casanova, G.; Matera, M.; Ruggiero, F.M.; Ferri, D.; Paradies, G. Mitochondrial dysfunction in rat with nonalcoholic fatty liver Involvement of complex I, reactive oxygen species and cardiolipin. Biochim. Biophys. Acta 2007, 1767, 1260–1267.
- Grattagliano, I.; Caraceni, P.; Calamita, G.; Ferri, D.; Gargano, I.; Palasciano, G.; Portincasa, P. Severe liver steatosis correlates with nitrosative and oxidative stress in rats. Eur. J. Clin. Investig. 2008, 38, 523–530.
- Grattagliano, I.; de Bari, O.; Bernardo, T.C.; Oliveira, P.J.; Wang, D.Q.; Portincasa, P. Role of mitochondria in nonalcoholic fatty liver disease--from origin to propagation. Clin. Biochem. 2012, 45, 610–618.
- Grattagliano, I.; Lauterburg, B.H.; Palasciano, G.; Portincasa, P. 13C-breath tests for clinical investigation of liver mitochondrial function. Eur. J. Clin. Investig. 2010, 40, 843–850.
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab. 2011, 14, 804–810.
- Schmid, A.I.; Szendroedi, J.; Chmelik, M.; Krssak, M.; Moser, E.; Roden, M. Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 2011, 34, 448–453.
- Peng, K.Y.; Watt, M.J.; Rensen, S.; Greve, J.W.; Huynh, K.; Jayawardana, K.S.; Meikle, P.J.; Meex, R.C.R. Mitochondrial dysfunction-related lipid changes occur in nonalcoholic fatty liver disease progression. J. Lipid Res. 2018, 59, 1977–1986.
- Koliaki, C.; Szendroedi, J.; Kaul, K.; Jelenik, T.; Nowotny, P.; Jankowiak, F.; Herder, C.; Carstensen, M.; Krausch, M.; Knoefel, W.T. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015, 21, 739–746.
- Fletcher, J.A.; Deja, S.; Satapati, S.; Fu, X.; Burgess, S.C.; Browning, J.D. Impaired ketogenesis and increased acetyl-CoA oxidation promote hyperglycemia in human fatty liver. JCI Insight 2019, 5.
- Grasselli, E.; Baldini, F.; Vecchione, G.; Oliveira, P.J.; Sardao, V.A.; Voci, A.; Portincasa, P.; Vergani, L. Excess fructose and fatty acids trigger a model of nonalcoholic fatty liver disease progression in vitro: Protective effect of the flavonoid silybin. Int. J. Mol. Med. 2019, 44, 705–712.
- An, P.; Wei, L.L.; Zhao, S.; Sverdlov, D.Y.; Vaid, K.A.; Miyamoto, M.; Kuramitsu, K.; Lai, M.; Popov, Y.V. Hepatocyte mitochondria-derived danger signals directly activate hepatic stellate cells and drive progression of liver fibrosis. Nat. Commun. 2020, 11.
- Grossini, E.; Garhwal, D.P.; Calamita, G.; Romito, R.; Rigamonti, C.; Minisini, R.; Smirne, C.; Surico, D.; Bellan, M.; Pirisi, M. Exposure to Plasma From Non-alcoholic Fatty Liver Disease Patients Affects Hepatocyte Viability, Generates Mitochondrial Dysfunction, and Modulates Pathways Involved in Fat Accumulation and Inflammation. Front. Med. 2021, 8.
- Reddy, J.K. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta-oxidation, PPAR alpha, and steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1333–G1339.
- Camporez, J.P.; Wang, Y.; Faarkrog, K.; Chukijrungroat, N.; Petersen, K.F.; Shulman, G.I. Mechanism by which arylamine N-acetyltransferase 1 ablation causes insulin resistance in mice. Proc. Natl. Acad. Sci. USA 2017, 114, E11285–E11292.
- Chennamsetty, I.; Coronado, M.; Contrepois, K.; Keller, M.P.; Carcamo-Orive, I.; Sandin, J.; Fajardo, G.; Whittle, A.J.; Fathzadeh, M.; Snyder, M.; et al. Nat1 Deficiency Is Associated with Mitochondrial Dysfunction and Exercise Intolerance in Mice. Cell Rep. 2016, 17, 527–540.
- Gijón, M.A.; Riekhof, W.R.; Zarini, S.; Murphy, R.C.; Voelker, D.R. Lysophospholipid Acyltransferases and Arachidonate Recycling in Human Neutrophils*. J. Biol. Chem. 2008, 283, 30235–30245.
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Boren, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.
- Luukkonen, P.K.; Zhou, Y.; Hyötyläinen, T.; Leivonen, M.; Arola, J.; Orho-Melander, M.; Orešič, M.; Yki-Järvinen, H. The MBOAT7 variant rs641738 alters hepatic phosphatidylinositols and increases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2016, 65, 1263–1265.
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448.
- Thabet, K.; Asimakopoulos, A.; Shojaei, M.; Romero-Gomez, M.; Mangia, A.; Irving, W.L.; Berg, T.; Dore, G.J.; Grønbæk, H.; Sheridan, D.; et al. MBOAT7 rs641738 increases risk of liver inflammation and transition to fibrosis in chronic hepatitis C. Nat. Commun. 2016, 7.
- Thangapandi, V.R.; Knittelfelder, O.; Brosch, M.; Patsenker, E.; Vvedenskaya, O.; Buch, S.; Hinz, S.; Hendricks, A.; Nati, M.; Herrmann, A.; et al. Loss of hepatic Mboat7 leads to liver fibrosis. Gut 2021, 70, 940–950.
- Helsley, R.N.; Venkateshwari, V.; Brown, A.L.; Gromovsky, A.D.; Schugar, R.C.; Ramachandiran, I.; Fung, K.; Kabbany, M.N.; Banerjee, R.; Neumann, C.K. Obesity-linked suppression of membrane-bound Oacyltransferase 7 (MBOAT7) drives non-alcoholic fatty liver disease. Elife 2019, 8, e49882.
- Fondevila, M.F.; Fernandez, U.; Gonzalez-Rellan, M.J.; Da Silva Lima, N.; Buque, X.; Gonzalez-Rodriguez, A.; Alonso, C.; Iruarrizaga-Lejarreta, M.; Delgado, T.C.; Varela-Rey, M.; et al. The L-α-Lysophosphatidylinositol/G Protein–Coupled Receptor 55 System Induces the Development of Nonalcoholic Steatosis and Steatohepatitis. Hepatology 2021, 73, 606–624.
- Petersen, K.F.; Befroy, D.; Dufour, S.; Dziura, J.; Ariyan, C.; Rothman, D.L.; DiPietro, L.; Cline, G.W.; Shulman, G.I. Mitochondrial dysfunction in the elderly: Possible role in insulin resistance. Science 2003, 300, 1140–1142.
- Lee, H.-Y.; Choi, C.S.; Birkenfeld, A.L.; Alves, T.C.; Jornayvaz, F.R.; Jurczak, M.J.; Zhang, D.; Woo, D.K.; Shadel, G.S.; Ladiges, W.; et al. Targeted Expression of Catalase to Mitochondria Prevents Age-Associated Reductions in Mitochondrial Function and Insulin Resistance. Cell Metab. 2010, 12, 668–674.
- Palmer, A.K.; Kirkland, J.L. Aging and adipose tissue: Potential interventions for diabetes and regenerative medicine. Exp. Gerontol. 2016, 86, 97–105.
- Lyu, K.; Zhang, Y.; Zhang, D.; Kahn, M.; ter Horst, K.W.; Rodrigues, M.R.S.; Gaspar, R.C.; Hirabara, S.M.; Luukkonen, P.K.; Lee, S.; et al. A Membrane-Bound Diacylglycerol Species Induces PKCϵ-Mediated Hepatic Insulin Resistance. Cell Metab. 2020, 32, 654–664.e655.
- Piccinin, E.; Villani, G.; Moschetta, A. Metabolic aspects in NAFLD, NASH and hepatocellular carcinoma: The role of PGC1 coactivators. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 160–174.
- Li, Y.; Chen, C.; Lu, L.; Guo, W.; VanWagner, L.B.; Shikany, J.M.; Zhang, S.; Kahe, K. Cadmium Exposure in Young Adulthood Is Associated with Risk of Nonalcoholic Fatty Liver Disease in Midlife. Dig. Dis. Sci. 2021.
- He, X.; Gao, J.; Hou, H.; Qi, Z.; Chen, H.; Zhang, X.X. Inhibition of Mitochondrial Fatty Acid Oxidation Contributes to Development of Nonalcoholic Fatty Liver Disease Induced by Environmental Cadmium Exposure. Environ. Sci. Technol. 2019, 53, 13992–14000.
- Frediani, J.K.; Naioti, E.A.; Vos, M.B.; Figueroa, J.; Marsit, C.J.; Welsh, J.A. Arsenic exposure and risk of nonalcoholic fatty liver disease (NAFLD) among U.S. adolescents and adults: An association modified by race/ethnicity, NHANES 2005–2014. Environ. Health Glob. Access Sci. Source 2018, 17, 6.
- Mozaffarian, F.; Dehghani, M.A.; Vanani, A.R.; Mahdavinia, M. Protective Effects of Alpha Lipoic Acid Against Arsenic Induced Oxidative Stress in Isolated Rat Liver Mitochondria. Biol. Trace Elem. Res. 2021.
- Chen, R.; Xu, Y.; Xu, C.; Shu, Y.; Ma, S.; Lu, C.; Mo, X. Associations between mercury exposure and the risk of nonalcoholic fatty liver disease (NAFLD) in US adolescents. Environ. Sci. Pollut. Res. Int. 2019, 26, 31384–31391.
- Pereira, L.C.; de Paula, E.S.; Pazin, M.; Carneiro, M.F.H.; Grotto, D.; Barbosa, F., Jr.; Dorta, D.J. Niacin prevents mitochondrial oxidative stress caused by sub-chronic exposure to methylmercury. Drug Chem. Toxicol. 2020, 43, 64–70.
- Qiu, Y.N.; Wang, G.H.; Zhou, F.; Hao, J.J.; Tian, L.; Guan, L.F.; Geng, X.K.; Ding, Y.C.; Wu, H.W.; Zhang, K.Z. PM2.5 induces liver fibrosis via triggering ROS-mediated mitophagy. Ecotoxicol. Environ. Saf. 2019, 167, 178–187.
- Breton, C.V.; Song, A.Y.; Xiao, J.; Kim, S.J.; Mehta, H.H.; Wan, J.; Yen, K.; Sioutas, C.; Lurmann, F.; Xue, S.; et al. Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion 2019, 46, 22–29.
- Chen, J.; Wu, L.; Yang, G.; Zhang, C.; Liu, X.; Sun, X.; Chen, X.; Wang, N. The influence of PM2.5 exposure on non-alcoholic fatty liver disease. Life Sci. 2021, 270, 119135.
- Khan, S.; Beigh, S.; Chaudhari, B.P.; Sharma, S.; Aliul Hasan Abdi, S.; Ahmad, S.; Ahmad, F.; Parvez, S.; Raisuddin, S. Mitochondrial dysfunction induced by Bisphenol A is a factor of its hepatotoxicity in rats. Environ. Toxicol. 2016, 31, 1922–1934.
- Huc, L.; Lemarie, A.; Gueraud, F.; Helies-Toussaint, C. Low concentrations of bisphenol A induce lipid accumulation mediated by the production of reactive oxygen species in the mitochondria of HepG2 cells. Toxicol. Vitr. Int. J. Publ. Assoc. BIBRA 2012, 26, 709–717.
- Liu, Q.; Wang, Q.; Xu, C.; Shao, W.; Zhang, C.; Liu, H.; Jiang, Z.; Gu, A. Organochloride pesticides impaired mitochondrial function in hepatocytes and aggravated disorders of fatty acid metabolism. Sci. Rep. 2017, 7, 46339.
- Wahlang, B.; Appana, S.; Falkner, K.C.; McClain, C.J.; Brock, G.; Cave, M.C. Insecticide and metal exposures are associated with a surrogate biomarker for non-alcoholic fatty liver disease in the National Health and Nutrition Examination Survey 2003-2004. Environ. Sci. Pollut. Res. Int. 2020, 27, 6476–6487.
- Miranda, C.A.; Guimaraes, A.; Bizerra, P.F.V.; Mingatto, F.E. Diazinon impairs bioenergetics and induces membrane permeability transition on mitochondria isolated from rat liver. J. Toxicol. Environ. Health A 2020, 83, 616–629.
- Rives, C.; Fougerat, A.; Ellero-Simatos, S.; Loiseau, N.; Guillou, H.; Gamet-Payrastre, L.; Wahli, W. Oxidative Stress in NAFLD: Role of Nutrients and Food Contaminants. Biomolecules 2020, 10, 1702.
- Vecchione, G.; Grasselli, E.; Voci, A.; Baldini, F.; Grattagliano, I.; Wang, D.Q.; Portincasa, P.; Vergani, L. Silybin counteracts lipid excess and oxidative stress in cultured steatotic hepatic cells. World J. Gastroenterol. WJG 2016, 22, 6016–6026.
- Nadanaciva, S.; Will, Y. New insights in drug-induced mitochondrial toxicity. Curr. Pharm. Des. 2011, 17, 2100–2112.
- Pereira, C.V.; Nadanaciva, S.; Oliveira, P.J.; Will, Y. The contribution of oxidative stress to drug-induced organ toxicity and its detection in vitro and in vivo. Expert Opin. Drug Metab. Toxicol. 2012, 8, 219–237.
- Masuo, Y.; Imai, T.; Shibato, J.; Hirano, M.; Jones, O.A.; Maguire, M.L.; Satoh, K.; Kikuchi, S.; Rakwal, R. Omic analyses unravels global molecular changes in the brain and liver of a rat model for chronic Sake (Japanese alcoholic beverage) intake. Electrophoresis 2009, 30, 1259–1275.
- Griffin, J.L.; Nicholls, A.W. Metabolomics as a functional genomic tool for understanding lipid dysfunction in diabetes, obesity and related disorders. Pharmacogenomics 2006, 7, 1095–1107.
- Malik, A.N.; Simoes, I.C.M.; Rosa, H.S.; Khan, S.; Karkucinska-Wieckowska, A.; Wieckowski, M.R. A Diet Induced Maladaptive Increase in Hepatic Mitochondrial DNA Precedes OXPHOS Defects and May Contribute to Non-Alcoholic Fatty Liver Disease. Cells 2019, 8, 1222.


