 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Amit K Tiwari | + 6598 word(s) | 6598 | 2021-06-28 11:05:25 | | | |
| 2 | Conner Chen | Meta information modification | 6598 | 2021-08-04 11:27:12 | | |
Video Upload Options
Chemotherapeutic drugs are primarily administered to cancer patients via oral or parenteral routes. The use of transdermal drug delivery could potentially be a better alternative to decrease the dose frequency and severity of adverse or toxic effects associated with oral or parenteral administration of chemotherapeutic drugs. The transdermal delivery of drugs has shown to be advantageous for the treatment of highly localized tumors in certain types of breast and skin cancers. In addition, the transdermal route can be used to deliver low-dose chemotherapeutics in a sustained manner. The transdermal route can also be utilized for vaccine design in cancer management, for example, vaccines against cervical cancer. However, the design of transdermal formulations may be challenging in terms of the conjugation chemistry of the molecules and the sustained and reproducible delivery of therapeutically efficacious doses.
1. Transdermal Delivery
Presently, the global share of transdermal products in pharmaceuticals is worth billions of dollars but is limited to a few drug molecules [1]. A majority of the commercial transdermal formulations are based on small molecules (molecular weight less than 500 Da) with moderate lipophilicity (log P between 1 and 4) [2]. The advantages of transdermal delivery include the ease of drug administration and termination, avoidance of injections and hospital visits, avoidance of drug degradation by gastric pH, enzymes and hepatic first-pass metabolism, and dose-related side effects. All of these advantages make the transdermal delivery a convenient and compliant administration route for patients [3][4][5].
The skin is the largest organ of the body, with a barrier property that is practically impermeable to many external chemicals, microbes, and particulate matters, including colloidal components [6]. Figure 1 depicts the schematic structure of human skin and major routes of transdermal drug transport (appendageal, transcellular, and intracellular routes). Due to the lipid rich nature of the stratum corneum (SC), the outermost layer of the skin, only those drugs with moderate lipid solubility can cross the SC. Even though there are some hydrophilic channels, and pores from the sweat ducts and hair roots, only the compounds with moderate aqueous and lipid solubility can permeate across the skin for topical and transdermal delivery [7]. The barrier property of the skin widely varies depending on the body site due to the varied thickness of SC, varied distribution of pores and hair roots, and age of the patient. Due to the varied barrier property, the transdermal products are specified for application at a particular body site, such as chest, thighs, under ears, underarms, or scrotum [8][9][10][11].
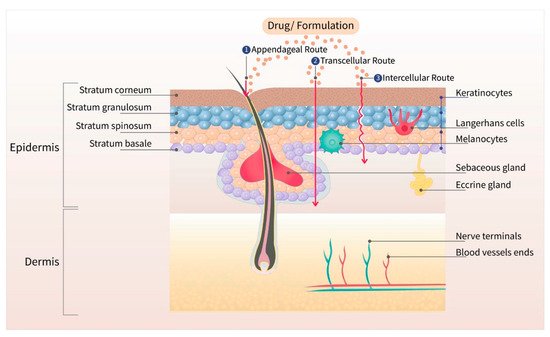
Figure 1. Different layers of the skin with three major routes (appendageal, transcellular, and intracellular) of drug transport. The outermost layer, stratum corneum, is the major barrier for transdermal drug delivery.
The transdermal permeation of a compound is dependent on its molecular size, partition coefficient (oil–water), and surface charge [12]. Based on the porosity of the SC, only those agents with a size less than 36 nm can diffuse through lipidic or aqueous channels [13]. Based on the follicular pore size, particulates below 100 nm accumulate in the sebaceous glands and hair follicles [14][15][16]. The transdermal drug candidate should have adequate solubility in the SC lipid bilayers, which is the rate-limiting step for drug absorption. Moreover, other factors, such as the melting point, molecular weight, or molar volume, also influence the permeation of drug across the skin [17][18]. To use the transdermal route, the drug candidate should have adequate skin permeability, should be potent enough to produce therapeutic drug concentration, and cause no skin sensitization or irritation. Only, a few drug molecules are suitable for passive transdermal drug delivery. Table 1 shows the ideal properties of a drug candidate for passive transdermal delivery. All the currently approved transdermal patches, creams, and liquids are based on drugs that possess these properties. Up to the present date, there are about twenty drug molecules that have been approved for transdermal administration [19]. None of the drugs belonging to the chemotherapeutic class has been approved for transdermal administration.
Table 1. Ideal physicochemical and pharmaceutical properties for passive transdermal drug delivery [2][20].
| Critical Properties | Ideal Limits |
|---|---|
| Aqueous solubility | >1 mg/mL |
| Lipophilicity (log octanol/water P) | >1 and <4 |
| Molecular weight | <500 Da |
| Melting point | <200 °C |
| pH of the saturated aqueous solution | 5–9 |
| Daily dose | <20 mg |
| Skin irritation or sensitization | None |
2. Transdermal Chemotherapeutics for Breast Cancer
2.1. Tamoxifen Citrate
Tamoxifen citrate is available in the form of solution (2 mg/mL as a base), and tablets (10 and 20 mg as a base) for oral administration. It is a trans-isomer of a triphenylethylene derivative, with a molecular weight of 563.6 Da. It acts as a selective estrogen receptor (ER) modulator that competes with β-estradiol for the alpha-estrogen (ERα), thus inhibiting the bioactivity of estrogen in breast tissue [21][22]. It is used (1) to treat estrogen receptor-positive (ER+ve) metastatic breast cancer; (2) as an adjuvant to treat patients with early stage ER+ve breast cancer; (3) to decrease the risk of invasive breast cancer after tumor excision and radiation in women with ductal carcinoma in situ (DCIS); and (4) to decrease the incidence of breast cancer in women at high risk [23][24]. The activity of the drug is due to the metabolites 4-hydroxytamoxifen (4-OHT) and N-desmethyl-4-hydroxytamoxifen (ENX), which are formed by the cytochrome P450 enzymes, CYP2D6 and CYP3A4/5, which have a higher affinity for ERα compared to tamoxifen [25][26].
Although the oral formulation significantly decreases the risk of recurrence of ER+ve DCIS, its use can achieve the following: (1) activate ERα receptors in the endometrium (increasing the risk of cancer); (2) increase the risk of venous thromboembolic events; (3) cause vasomotor symptoms and vaginal symptoms such as dryness, discharges, and atrophy [27][28][29]. Furthermore, the efficacy of oral tamoxifen could be decreased in women with polymorphisms in CYP2D6 and CYP3A4/5 as this would decrease the levels of tamoxifen’s active metabolites [25][26]. The topical delivery of 4-OHT gel to the local tumors was shown to be a suitable alternative for oral delivery, decreasing the risk of systemic adverse effects. A randomized, pre-surgical trial in pre- and post-menopausal women was conducted by comparing transdermal 4-OHT gel (4 mg/day) to the oral tamoxifen (20 mg/day) [27]. The results indicated that equivalent concentrations of 4-OHT, i.e., 5.4 and 5.8 ng/g, were obtained in breast tissue biopsy samples from patients treated with oral and transdermal formulation, respectively. There was no significant correlation between the amount of 4-OHT in the tissue and the plasma in the transdermal formulation treated group. Thus, using the transdermal route sufficient amount of drug can be delivered to the target tissue without spiking the 4-OHT concentration in the blood. In contrast, with the oral tamoxifen group, there was a significant correlation between the concentration of 4-OHT in the plasma and the tissue leading to a spike in drug concentration, thereby causing systemic side effects. Therefore, the transdermal 4-OHT gel was found to be superior when compared to oral tamoxifen in localizing 4-OHT in patients with DCIS, thus decreasing the incidence of adverse systemic side effects, and thereby increasing the patient compliance [27]. In other study, Pathan et al. optimized a nano-emulsion of tamoxifen citrate by using arachis oil, Cremophore EL, and ethanol [30]. The solubility of tamoxifen was highest in arachis oil compared to other oils such as jojoba oil, coconut oil, castor oil and sesame oil. Among different surfactants and co-surfactants (Labrafil, Tween-80, Cremopore EL, ethanol, butanol, and propanol), Cremophore EL and ethanol demonstrated a better solubility profile for tamoxifen citrate. The optimized nano-emulsion possessed surfactant to co-surfactant ratio of 1:1, surfactant to oil ratio of 1:9, and a drug content of 5% w/w. This optimized formulation displayed a mean droplet size of 68 nm with a polydispersity index (PDI) of 0.125, and viscosity of 201 cP. The flux of tamoxifen evaluated by using Keshary–Chien diffusion cells across the excised rat skin was found to be 98.98 µg/cm2/h [30]. However, a control formulation of tamoxifen was not used to compare the efficacy of the formulation. Lin et al. [31] used bioceramic irradiation, with an emissivity of 0.98 at a wavelength of 6 to 14 μm, to enhance the transdermal delivery of tamoxifen. The permeation across cellulose acetate membrane showed that the bioceramic irradiation on water molecules weakens the hydrogen bond, which decreases the viscosity, thereby enhances the permeation. The study demonstrated higher permeation of tamoxifen and indomethacin by using bioceramic irradiation compared to the control, where bioceramics were not used [31]. Similarly, Lee et al. [32] evaluated the relative efficiency of the skin permeation of 4-OHT and ENX, using split-thickness human skin. The permeation of ENX across human skin was improved by using oleic acid as a permeation enhancer. However, the permeation of 4-OHT was significantly lower than that of ENX, even in the presence of oleic acid [32]. Dendrimer-based micelles were prepared by Yang et al. [33] to determine the feasibility of delivering ENX by the transdermal route. Generally, dendrimer-based drug-delivery systems involve the chemical conjugation of the drug with the surface groups of dendrimers to increase the drug stability during transport [34]. Due to a limited number of reactive functional groups on ENX, dendrimer conjugation was not feasible. Therefore, PEGylated dendro-based copolymers (PDCs) that can self-assemble into dendron micelles (DM) were utilized for ENX delivery [33]. The solubility and sequestration of ENX in DM was higher (3% drug loading efficiency, mean size of 48.4 nm), with smaller and uniform particle size distribution compared to the ENX cationic liposomes (0.1% drug loading efficiency, mean size of 100 nm) made with DOTAP, DMPC and cholesterol at a molar ratio of 2:2:1. Furthermore, cell titer 96 aqueous one solution (MTS) assay in ER+ve MCF-7 and ER-ve MDA-MB-231 breast cancer cell lines indicated that the ER-dependent, anti-proliferative efficacy of ENX was retained after encapsulation in DMs. In contrast to the liposomal formulation, the permeation of ENX from DMs across rat and human skin were sustained over 6 days. However, the intactness of the skin at the end of the study was not demonstrated. The flux of the ENX across the skin from DMs was proportional to the sequestered drug in the DMs. Despite poor drug loading, the liposomes demonstrated the highest permeability coefficient (Kp 0.0467 cm/h). Moreover, the DMs (size >24 nm) cannot permeate through the aqueous pores of the skin (size <4 nm), as they have a significantly larger size compared to the aqueous pores of the skin. However, DMs facilitate the permeation of drugs through the skin by translocating the drug molecules [33].
Lin et al. [35] used the liposome–PEG–PEI complex (LPPC) for the transdermal delivery of tamoxifen in vivo in mice xenograft with BT474 breast cancer cells. The in vitro efficacy of the LPPC-tamoxifen was determined in the breast cancer cell lines, ER+ve MCF-7, DT474 and ER-ve MDA-MB-231, using the 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT assay). Although the IC50 values were not determined, the liposomal formulation significantly reduced the viability of the ER+ve and ER-ve cell lines. Tamoxifen arrested the proliferation of ER-ve breast cancer cell lines in the S phase of the cell cycle. Furthermore, the study showed that the inhibition of the CIP2A/PP2A/p-Akt (Protein phosphatase 2A (PP2A), a cellular inhibitor of PP2A/CIPP2A, protein kinase B-Akt) signaling pathway was responsible for the inhibition of ER-ve cell proliferation. The LPPC-tamoxifen (LPPC/TAM) decreased tumor growth by 82% compared to the control. LPPC/TAM was also found to effectively inhibit BT474 tumor growth than cream-tamoxifen. In addition, the tissue damage and pathology of organs induced by LPPC/TAM treatment were assessed. The results in the treated mice showed that LPPC/TAM did not cause any skin irritation or injury to organs (Figure 2). Overall, the results indicated the feasibility of using LPPC formulations for the local delivery of tamoxifen in breast cancer cells in an in vivo mouse model [35]. Several research studies demonstrated that local delivery of tamoxifen and its derivatives via transdermal route is possible. Moreover, the compounds disperse in a gel matrix or in the form of nanomedicines (ethosomes, liposomes, dendrimers, etc.) and can provide effective drug levels at the tumor sites in animal models.
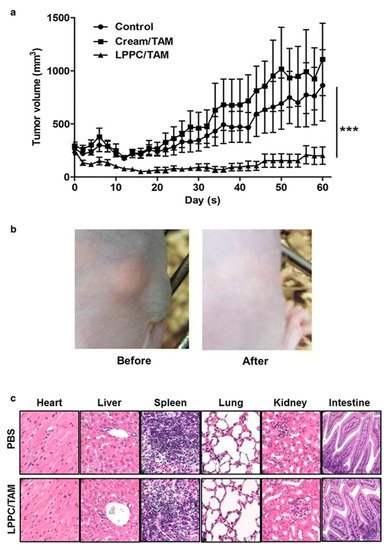
Figure 2. Antitumor efficacy of liposome–PEG–PEI complex/Tamoxifen (LPPC/TAM) in BT474-tumor-bearing mice via transdermal treatment. (a) Following transdermal application of the cream/TAM or LPPC/TAM to the tumor area every day, the tumor volume was measured with a caliper, and was calculated as L × H × W × 0.5236. The animals were sacrificed after 60 days of implantation of the 60-day release 17β-estradiol pellet (n = 5). (b) Observations of the skin in the tumor-bearing mouse before and after treatment with LPPC/TAM. (c) Histopathological evaluations of the heart, liver, spleen, lungs, kidneys, and intestines in the LPPC/TAM treatment group. Sections of the tissue were fixed in 10% formaldehyde overnight, embedded in paraffin and were cut into slices. Later, tissue sections were stained by using H&E. *** p < 0.001. Reproduced with permission from Lin et al. [35].
2.2. Letrozole
Letrozole is available as 2.5 mg tablets for oral administration. It is a dibenzo nitrile derivative, which has a molecular weight of 285.3 Da and a log P of 1.27. It is used in the treatment of estrogen-dependent breast cancer. It is a reversible inhibitor of the enzyme aromatase, which catalyzes the last step in the synthesis of estrogen, thereby blocking the production of estrogen [36][37]. The oral formulation of letrozole significantly decreases the plasma levels of estrogen [36]. However, estrogen levels in local tissue, such as the breast, can be significantly greater than in the plasma [36][38][39]. Furthermore, the depletion of circulating estrogens by letrozole can produce vasomotor symptoms and adverse effects on the bone tissue [38][39].
The physicochemical properties and the low dose of letrozole are very favorable for designing a transdermal formulation. Li et al. [40] determined the concentration of letrozole in plasma, skin, and breast tissue of mice that received oral suspension (50 mg/kg) or transdermal patch containing 3 mg/5cm2 of letrozole. Following the oral administration, letrozole level in breast tissue and plasma were 0.15–2.38 μg/g and 0.20–4.80 μg/mL, respectively, whereas, after transdermal administration, the letrozole level was 10.4–49.3 μg/g, 1.6–6.8 μg/g, and 0.35–1.64 μg/mL in the skin, breast tissue, and plasma, respectively. The oral delivery showed elevated plasma concentration of letrozole compared to the skin. Overall, the transdermal delivery of letrozole was more efficient, which showed localized delivery to the breast tissue instead of elevating the plasma concentration of the drug. Moreover, the low systemic levels of letrozole from the patch could decrease the incidence of the aforementioned adverse effects [40]. Li et al. [41] optimized the delivery from the transdermal patch of letrozole by using various adhesives, permeation enhancers, and different letrozole concentrations. The permeation of letrozole through excised rat skin was significantly higher through the patch with adhesive DURO-TAK® 87-4098, which lacks a carboxyl group, compared to DURO-TAK® 87-2677 and 87-2852, the adhesives with a carboxyl group. It was speculated that the triazole group of letrozole could interact with the carboxyl group to form hydrogen bonds [41], which could impede the release of the drug from the adhesive with carboxylic groups. Conventional chemical enhancers were used to optimize the permeation, and the results indicated that a combination of azone (10%) with propylene glycol (5%), containing 1.5% of letrozole, was optimal for the adhesive patch [41]. Maniyar et al. [42] formulated spray-dried letrozole (SPD-LET) as a liposomal dispersion in cream for topical delivery to breast cancer tumors. The liposomal cream, which contained peppermint oil, produced a greater permeation than olive oil. In vitro experiments using MTT assay indicated that the SPD-LET formulation had superior anti-proliferative activity with lower viability in MDA-MB-231 (breast cancer) cell line compared to the group treated with plain letrozole cream. Moreover, the in vivo pharmacokinetic profile in Wistar rats was compared between the plain letrozole cream and SPD-LET cream. The Cmax, Tmax, and AUC were 11.3 μg/mL, 3 h, and 101.7 μgh/mL for SPD-LET cream, whereas 4.2 μg/mL, 5 h and 37.8 μgh/mL for plain letrozole cream. Overall, the pharmacokinetic profile of the letrozole liposomal cream was superior to that of plain letrozole cream. It was assumed that the SPD liposomes contain lipid components that get adsorbed on the SC and progressively merge into the polar lipids which enhances the drug delivery across the skin [42]. The existing literature suggest that letrozole formulated in the form of a transdermal patch or a liposomal cream are effective to deliver the drug across the skin into the local tumor tissue, and into the systemic circulation. Since letrozole tablets induce nausea, vomiting, and hot flashes, transdermal administration could potentially be an alternative to overcome the side effects.
2.3. Anastrozole
Anastrozole is available as 1 mg tablets for oral administration. It is a benzenediacetonitrile derivative and has a molecular weight of 293.4 Da. It binds to the cytochrome P-450 component of aromatase and reversibly inhibits its activity [43]. Although anastrozole and letrozole belong to the same class of aromatase inhibitors (triazoles that are reversible aromatase inhibitors), they differ in their pharmacological profile (letrozole seems to be more potent compared to anastrozole) [43]. Similar to letrozole, the physicochemical and pharmacological properties of anastrozole are very favorable for designing a transdermal formulation. Xi et al. [44] designed an adhesive matrix transdermal patch of anastrozole, using DURO-TAK® 87-4098, adhesive without the carboxylic group, and 8% isopropyl myristate (IPM). Anastrozole also contains a triazole moiety as letrozole, which is a hydrogen bond donor and acceptor. It was observed that DURO-TAK® without carboxylic groups showed higher drug delivery (3.84 times higher) compared to the DURO-TAK® adhesive patch with a carboxylic group. Among the different penetration enhancers evaluated (i.e., Transcutol®, IPM, oleic acid, and l-menthol), IPM at a concentration of 8% produced the highest flux (26.13 ± 6.75 μg/cm2/h) of anastrozole across the excised rat abdominal skin. An in vivo study was conducted in mice comparing the oral suspension (15 mg/kg) and transdermal patch (2 mg/cm2) of anastrozole, where the concentration of drug in skin, muscles, and plasma were quantified at different time points. Anastrozole delivered by transdermal route localized in the skin and muscles as a local depot providing sustained plasma level for 12 h, whereas the oral anastrozole was absorbed systemically and reached a peak plasma concentration within 1 h. The muscle–plasma concentration ratios were 49.06, 43.02, 26.91, 41.48, and 51.29 at a time point of 0.17, 1, 4, 8, and 12 h via transdermal route while the ratios were 0.79, 077, and 1.09 in 0.17, 1, and 4 h via oral route. The results indicated that a sustained delivery of anastrozole from the patch to the skin and surrounding tumors could be achieved [44]. Regenthal et al. [45] formulated a transdermal patch of anastrozole and compared its pharmacokinetic profile in dogs with the PK results obtained by Mende et al. [46], using the oral anastrozole formulation (tablets) in human subjects. Silicon matrix BIO-PSA® type 7-4302, ethyl acetate as a solvent, glycerol as crystallization inhibitor, and CoTran™ 9720 backing film were used in the transdermal patch. The transdermal patch produced a rapid and linear delivery of anastrozole in beagle dogs within the first 24 h (Cmax = 5.8 ng/mL), and after reaching a plateau, there was a slow decrease in the next two days [45]. The oral tablets of anastrozole produced a rapid, maximal concentration, whereas the transdermal patch produced a steady release of the anastrozole. The area under the curve (AUC) for the transdermal anastrozole patch was comparable to the oral formulation, but the half-life of anastrozole increased 2-fold following the application of the transdermal patch [45]. The state of the drug in the patch (i.e., homogeneity and crystallinity) significantly affects the drug-release pattern from the patch. It was also shown that ethyl acetate was superior over other solvents such as THF, DMSO, xylene, ethanol, dioxane, chloroform, and dichloromethane, owing to its higher drug solubility and compatibility with the adhesives [45]. Unlike oral formulations of anastrozole the transdermal patch does not cause a spike in the plasma drug levels, thereby avoiding the adverse effects associated with it.
3. Transdermal Chemotherapeutics for Melanoma
3.1. Imatinib Mesylate (IM)
Imatinib mesylate (IM) is a benzamide methanesulfonate derivative, which has a molecular weight of 589.7 Da. It is available as tablets (100 mg and 400 mg) for oral administration. IM is used to treat patients with certain types of chronic myelogenous leukemia (CML). It binds in an area in the BCR-ABL protein that binds its substrate, ATP, and inhibits the catalytic transfer of a phosphate group to a specific tyrosine, thus inhibiting the phosphorylation of certain proteins that mediate the pathophysiology of CML. In addition, imatinib inhibits the protein, c-kit, a Type III receptor tyrosine kinase (also known as CD117) that is activated by the endogenous proteins, stem cell factors (SCFs), present in certain types of tumors [47]. The overexpression of c-kit receptors in melanoma increases the expression of the microphthalmia-associated transcription factor and downregulates the anti-apoptotic protein, Bcl2, thereby promoting cancer progression [48].
Although IM is highly lipophilic (log P 4.38), and has a high dose, researchers have attempted transdermal formulations for this drug by using carrier systems such as nanoparticles and active permeation techniques. Gold nanoparticles (AuNP) have been utilized for the delivery of different anticancer molecules, such as cetuximab, gemcitabine, cisplatin, etc., primarily by conjugating the drug to the surface of the particles [49]. Filon et al. have used both intact and damaged human skin to demonstrate the route of delivery of nanoparticles following transdermal application is typically paracellular or appendageal [50][51]. For the delivery of IM, Labala et al. [52] used positively charged polymers, polyethylene imine (PEI), and polystyrene sulfonate (PSS), to coat the gold nanoparticles by using a layer-by-layer strategy that produced stable gold nanoparticles without a significant increase in the particle size. IM was loaded on gold nanoparticles for active transdermal delivery, i.e., iontophoretic transport system. Moreover, the typical features of layer-by-layer polymer-coated AuNPs, such as small particle size (98.5 nm), high surface-charge density, and positive charge (32.3 mV), in combination with iontophoresis (0.47 mA/cm2 for 4 h), make them a suitable candidate for permeation across the skin [52]. The loading efficiency of IM was 28%, which was most likely released via a diffusion-controlled mechanism from the NPs. The permeation of IM-loaded AuNPs in conjunction with iontophoresis was 6.2-fold higher compared to the passive application. Moreover, in the cytotoxicity assay, the IM containing nanoparticles produced 80% inhibition of B16F10 melanoma cells at a concentration of 77.5 μM [52]. However, the formulations containing PEI should take cytotoxicity of PEI into account which could be associated with its high density of cationic charge and can also vary depending on the cell line [53]. In another similar study, Labala et al. [54] used a combination anticancer-drug approach, consisting of signal transduction and activator of transcription factor 3 (STAT3) siRNA with IM in AuNPs coated with multiple layers of chitosan. The chitosan provides a layer of positive charge, where the negatively charged siRNA could be sandwiched, and IM encapsulation solely depended on electrostatic interactions and hydrogen bonding, which enhanced the efficiency of drug loading. The dual drug-loaded nanoparticles, at 130 µM, significantly inhibited the growth of B16F10 melanoma cells in vitro. The in vivo efficacy of the nanoparticles was determined by using C57BL/6 mice with B16F10 melanoma cells via transdermal iontophoresis and intra-tumoral routes. The reduction of the tumor weight and volume following treatment via intra-tumoral route and transdermal route (iontophoresis: 0.5 mA/cm2 for 2 h) were not statistically significant. This result demonstrated comparable efficacy between the iontophoretic treatment and the intra tumoral delivery. To validate the molecular basis of successful delivery of the STAT3 siRNA, the levels of STAT3 expression were determined by using Western blots. The expression levels of STAT3 were significantly decreased with the transdermal (iontophoretic) administration of siRNA-IM AuNps compared to control. However, the drug loading in NPs must be further improved to ensure maximal delivery by using minimal NPs. The use of other human skin cancer cell lines, along with molecular markers indicative of cytotoxicity, would help to further validate the development of such novel delivery systems [54]. In summary, IM showed promising results in a nanoparticle based transdermal formulation. These studies indicate the possibility to deliver molecules via transdermal route even if the molecule needs to be delivered at a higher dose or has high lipophilicity.
3.2. Vemurafenib
Vemurafenib is a propane sulfonic acid derivative and has a molecular weight of 489.9 Da. It is available as tablets (240 mg) for oral administration. It is an inhibitor of the kinase activity of the BRAF kinase that has the valine to aspartate mutation at position 600 (i.e., the BRAF(V600E) kinase) [55], which is present in at least 60% of all melanomas [56]. Clinical studies indicate that the oral formulation of vemurafenib can produce hepatic and renal toxicity [57][58][59][60]. Vemurafenib is specifically approved for the treatment of metastatic melanoma. Zou et al. [60] evaluated the efficacy of the transdermal vemurafenib delivered via peptide-modified liposomes, using an in vivo mouse xenograft model. The peptide TD (ACSSSPSKHCG) was used as a permeation enhancer that transiently opens the paracellular pathway of the skin, thereby facilitating drug permeation [61]. To assess the toxicity profile of the liposomal content, the viability of A375, B16F10, and HUVEC cells were determined after treating those cell lines with blank liposomes for 72 h followed by MTT assay. Viability greater than 95% was reported which indicated the non-toxic nature of liposomal content and permeation enhancer. The in vitro permeation study carried out by using rat abdominal skin showed significantly higher permeation of vemurafenib from peptide-modified liposomes compared to liposomes without TD. For safety study BALB/c mice were given liposomal formulation every two days, for 7 days, at a dose of 0.25 mg of vemurafenib via three different routes, i.e., tail vein, oral gavage, and transdermal (abdominal region). Histopathological evaluation of kidney, heart, lungs, and liver showed significant damage to liver, lungs, and kidney in animals treated via oral and intravenous route, but such toxicity was not observed in the group treated via transdermal route. The in vivo study was carried out using xenograft model (BABL/c nude male mouse injected subcutaneously with BRAF mutant A375 cells) in which the liposomes containing 0.25 mg of vemurafenib was administered every 2 days, for 18 days, via three different routes oral, tail vein, and transdermal (daubed at the site of tumor). The tumor weight and tumor volume suppression in the group treated via transdermal route were much smaller compared to the other routes (oral and intravenous routes) [60]. This study exemplifies the possibility to overcome the oral toxicity of vemurafenib by using transdermal administration as an alternative route. Even though the oral dose is high, vemurafenib can still be delivered topically to the tumor regions to avoid undue systemic exposures.
3.3. Five-Aminolevulinic Acid (5-ALA) Hydrochloride
Chemically, 5-Aminolevulinic acid (5-ALA) hydrochloride is 5-amino-4-oxo-pentanoic acid hydrochloride with a molecular weight of 167.59 Da. It is available in the form of a lyophilized powder for oral solution (30 mg/mL), topical gel (10%), and topical solution (20%). Moreover, 5-ALA is an FDA-approved photodynamic therapy (PDT)—based drug used for the targeted therapy of cutaneous T-cell lymphoma, basal cell carcinoma, and squamous cell carcinoma [62][63]. PDT is based on the biotransformation of a prodrug to its active form, using light irradiation [64]. Moreover, 5-ALA is bio-transformed to protoporphyrin IX (PpIX) following irradiation with light, at a wavelength of 635 nm, which induces the formation of reactive oxygen species, resulting in tumor cell death [65]. Although 5-ALA is a small molecule, its permeation across the skin is limited by its hydrophilic nature. In order to overcome this limitation, Pierre et al. [66] prepared liposomes of size 400 nm, consisting of ceramides (50%), cholesterol (28%), palmitic acid (17%), and cholesteryl sulfate (5%), that had a similar composition to the mammalian SC to increase the delivery of 5-ALA across the skin. As expected, only 5.7% of the drug was encapsulated in the liposomes as 5-ALA is very hydrophilic in nature. The in vitro permeation study was carried out by using freshly excised rat dorsal skin over 36 h. The total amount of drug permeated and flux across the skin was higher for aqueous solution (3681 ± 104.65 μg and 38.3 ± 2.4 μg/cm2h) compared to the liposomal formulation (500.9 ± 32.5 μg and 4.2 ± 0.2 μg/cm2h). However, the amount of 5-ALA retained in the dermis and the epidermal layer was significantly higher for the liposomal formulation of 5-ALA compared to the control (5-ALA solution). Thus the liposomal formulation helps in localizing the drug around the application site [66]. Lin et al. [67] used 1,2 dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) to produce a liposomal formulation of 5-ALA to further increase the entrapment efficacy of the drug in carrier systems to 15–16%, where the liposome size was 100 nm. In vitro, cytotoxicity assay was carried out in B16F10 melanoma cells, where a light dose of 50 J/cm2 was used over 20 min after liposomal treatment. The viability of the cells was 52% after treatment with 5-ALA liposomes without DPPC whereas the viability was 33% with 5-ALA-DPPC liposomes. Further, the mitochondrial membrane potential was significantly reduced, and intracellular ROS levels significantly increased in 5-ALA-DPPC-treated cell lines compared to the 5-ALA liposomes without DPPC. In a mouse xenograft tumor model (B16F10 cell implanted subcutaneously), the tumor volume was significantly smaller in those treated with liposomes of 5-ALA compared to control. However, there was no significant difference in tumor volume in 5-ALA liposome treated, and 5-ALA/DPPC-treated group. Nevertheless, the level of Protoporphyrin IX (PpIX) in the tumor was significantly higher in 5-ALA/DPPC liposome treated group compared to the 5-ALA liposome group indicating the DPPC enables better permeation of drug across the skin to reach the tumor site [67]. In summary, 5-ALA is a hydrophilic drug that has been used for PDT and researchers made attempts to deliver this drug to the tumor sites as a liposomal formulation. Due to poor entrapment efficiency of the liposomes, and the larger dose requirements, currently, there are no effective formulations for topical delivery of 5-ALA. Ethosomes and niosomes are promising delivery systems for hydrophilic compounds [68] which could serve as potential carriers to deliver 5-ALA across the skin to the tumor sites.
4. Plant Product–Based Transdermal Chemotherapeutics
4.1. Curcumin
Curcumin is a polyphenolic constituent of turmeric powder that has various health benefits and antitumor efficacy [69]. It has been postulated that curcumin’s anticancer efficacy could be due to the induction of apoptosis, inhibition of of certain intracellular transcription factors and the downregulation of various secondary messengers, COX2, c-Jun, nitric oxide synthase, and matrix metalloproteinase-9 [70]. Curcumin is poorly soluble in water leading to poor absorption, which is one of the reasons for its low oral bioavailability. The other reasons include high instability, rapid metabolism, and rapid systemic elimination; all of these contribute to low oral bioavailability [71]. Lee et al. [72] summarized the nano-formulation strategy for curcumin and the limitations related to its use as an anticancer therapy. Here, we primarily focus on discussing recent publications that specifically report using transdermal formulations for the delivery of curcumin. Sun et al. [73] used hydroxypropyl-β-cyclodextrin (HP-β-CD)-curcumin complexation approach to improve the solubility and stability of curcumin. The grinding method was used for complexation where the molar ratio of curcumin to HP-β-CD was 1:2. An inclusion efficacy of 97.4% was achieved. The complex was further formulated as a hydrogel with poloxamers 407 and 188. This study demonstrated a 20-fold increase in the water solubility of curcumin along with higher photostability. The inclusion complex preserves the labile phenol hydroxyl group from degradation. The viability of B16F10 cells after the treatment with curcumin hydrogel (300 μg/mL) and curcumin-inclusion-complex hydrogel (300 μg/mL) was 83.1% and 15.12% respectively. The higher efficacy of the curcumin inclusion hydrogel was attributed to the enhanced solubility of the inclusion complex in the hydrogel [73]. Jose et al. [74] used deformable cationic liposomes in combination with iontophoresis to co-deliver curcumin and STAT3 siRNA. STAT3 is an oncogenic transcription factor that is overexpressed in various types of cancer, including melanoma [75]. Its transcriptional activity can be inhibited by small interference RNA (siRNA) [76]. However, there are significant challenges associated with the delivery of siRNA due to its poor in vivo stability and penetration across the cell membrane barrier [77]. The uptake of liposomes was studied in A431 cells with or without the endocytosis inhibitors (Chlorpromazine hydrochloride and methyl-β-cyclodextrin). The uptake was reduced when the inhibitor was used compared to the cells without the endocytosis inhibitor indicating the uptake occurred via Clathrin and caveolae pathway. MTT assay was carried in A431 cells where the combination of STAT siRNA and curcumin liposomes (with 250 µM curcumin and 0.5 nM STAT siRNA) showed highest growth inhibition of 72.9% compared to curcumin liposomes (350 µM) and SiRNA liposomes (1 nM), which showed inhibition of 32.2% and 56.9% respectively. Further iontophoresis, at a current density of 0.47 mA/cm2 for 4 h, was used to enhance the permeation of liposomes across the porcine ear skin. The group demonstrated 5-fold higher deposition of the curcumin in the skin by using iontophoresis compared to passive permeation of the liposomes suggesting transdermal delivery of the complex is feasible [74].
Several studies suggest that curcumin could target different pathways associated with breast cancer, which can be useful in the treatment of certain types of breast cancer [78]. Recently, studies have been conducted to develop transdermal delivery of curcumin to the breast tissue. Atlan et al. [79] proposed the fabrication of disposal bra inserts for the transdermal delivery of curcumin. Since curcumin is clinically safe up to doses of 8 g/day [79], they proposed the use of transferosomes to deliver curcumin in bra inserts as a preventive measure against breast cancer. The authors postulated that a preventive regimen of curcumin would modulate inflammatory biomarkers, limit ROS-induced damage to existing breast cells and eliminate incipient abnormal cells before they proliferate, thereby reducing the incidence of breast cancer [79]. Abdel-Hafez et al. [80] evaluated the effect of the penetration enhancers (Labrasol®, Transcutol®, limonene, and oleic acid) on the permeation of curcumin transferosomes by using phosphatidylcholine across the dorsal and abdominal excised skin of mice. The flux of curcumin from the oleic acid (15.058 μg/cm2h) and Transcutol® (15.678 μg/cm2h) formulations was higher compared to the flux of curcumin from Labrasol® (10.266 μg/cm2h) and limonene (10.189 μg/cm2h) [80]. Pushpalatha et al. [81] formulated nano-sponges of cyclodextrin, using pyromellitic dianhydride as a crosslinker. The nano-sponges were loaded with curcumin and resveratrol and were dispersed in a carbopol gel for transdermal delivery. The permeability study across porcine ear skin indicated that the hydrogel with nano-sponges produced a 10-fold and 2-fold higher permeation of curcumin and resveratrol, respectively, compared to hydrogel formulation without nano-sponges. The nano-sponge formulation also produced a 7-fold increase in the photostability of curcumin and resveratrol compared to hydrogel without nano-sponges. MTT assay in MCF-7 cells, treated with a combination of curcumin and resveratrol nano-sponges in the ratio of 1:1 and 1:3 resulted in IC50 of 15 µg/mL and 10 µg/mL, respectively [81]. All of these studies indicate that a suitable formulation design not only enhances the transdermal permeation but also promotes drug stability especially of those drugs, which are photosensitive such as curcumin.
4.2. Resveratrol
Resveratrol is a phenolic antioxidant present in natural foods, such as grapes, wine, berries, nuts, etc. [82]. Numerous studies suggest that resveratrol has anticancer activity in skin, breast, lung, liver, prostate, colon, and ovarian cancers [83]. However, the pharmacokinetic profile of resveratrol is not ideal for therapeutic use. Resveratrol is well absorbed orally by passive transport (~75%), but it undergoes extensive hepatic metabolism by glucuronidation and sulfate conjugation. In addition, resveratrol forms complexes with the low-density lipoproteins, plasma proteins such as albumin, leaving less than 1% resveratrol in the systemic circulation [84]. Therefore, there is a need for the development of bioavailable and efficacious formulations of resveratrol.
Resveratrol, by virtue of its antioxidant properties prevents UV-induced skin damage. In the skin, it inhibits lipid peroxidase and activation of NFkB [85]. Resveratrol skin treatment pre- and post-exposure to UVB light dramatically reduced the skin damage and skin cancer occurrence [86]. In addition to chemo-preventive effects, resveratrol also inhibits tumor progression by suppressing the growth of skin cancer by inhibiting DNA polymerase and deoxy-ribonucleotide synthesis and inducing cell-cycle arrest [87]. Tsai et al. [88] optimized and evaluated the potential of nanostructured emulsion carriers composed of isopropyl myristate or caproyl 90 with different surfactants (Brij 35, Tween 80, and L44) for transdermal delivery of resveratrol. IPM was used as an oily phase. The optimized nanostructured emulsion (size 277 nm and viscosity 5.82 cps) contained IPM (HLB 11.1) with Tween80/Span20 (with an HLB value of 11.16). The skin permeation and deposition of resveratrol across excised rat skin at the end of 24 h from saturated aqueous resveratrol solution (control) were 0.79 ± 0.78 and 4.26 ± 0.58 μg/cm2 whereas it was 276 ± 42.3 and 29.4 ± 7.7 μg/cm2 when optimized nano-emulsion consisting IPM with Tween80/Span20 was used. There was an 896.2-fold increase in drug permeation and a 10.2-fold increase in skin deposition with the use of nano-emulsion. Bioavailability experiments indicated that orally administered resveratrol suspension (dose 30 mg/kg) was rapidly metabolized and eliminated from the blood within 10 h of administration. In contrast, the transdermal administration of resveratrol (dose: 67 mg/kg applied on the shaved abdomen) produced a steady and prolonged level of resveratrol in the blood with Cmax around 25 h [88]. Similarly, Hu et al. [89] used a non-aqueous, self-double-emulsifying drug delivery system (SDEDDS) for transdermal delivery of resveratrol. SDEDDS is based on the principle that the drug is highly soluble in the innermost oily layer but less soluble in the outer oily layer, followed by the layer of surfactants [90]. Solubility of resveratrol in different organic phases (PEG400 < Transcutol® CG < propylene glycol < ethanol) and natural oils were reported (olive oil < evening primrose oil < aloe oil < avocado oil < grape seed oil < soybean oil < corn oil < coconut oil.) Evening primrose oil, Polyglycerol polyricinoleate (PGPR) as a hydrophobic surfactant (8%), Tween 60 (4%) as a hydrophilic surfactant and organic phase constituting PEG400, propylene glycol, and Transcutol® were used in the optimized formulation. The optimized formulation produced a biphasic release of the resveratrol, i.e., a burst release followed by a controlled release. The biphasic release was due to distribution of the drug between the o/o emulsion and the hydrophilic surfactant during homogenization. Although resveratrol was initially solubilized in the innermost oily layer, the drug is distributed in the outer most layer of the hydrophilic surfactant during homogenization. The burst release of resveratrol is due to the release of the drug from the outer layer. In contrast, resveratrol in the inner oily layer was released in a sustained manner. In vitro permeation across porcine ear skin showed 8.3 fold higher flux and 10 folds higher skin deposition compared to the aqueous solution of resveratrol [89]. Park et al. [91] used chitosan-coated liposomes to enhance the skin permeation of resveratrol. An in vitro permeation across the full thickness of dorsal mouse skin with chitosan-coated liposome (containing 0.1% resveratrol) showed a 126.93 μg/cm2 (40.42%) permeation of resveratrol over 24h. The uncoated liposomes showed 96.85 μg/cm2 (30.85%) permeation. Chitosan imparts a positive charge to the liposomes, which interact with the negative charge of the SC, thereby facilitating the permeation of the drug carrier system [91]. Pentek et al. [92] developed a dendrimer–resveratrol complex by using fourth generation polyamidoamine (PAMAM) dendrimers. This formulation increased the solubility and stability of resveratrol in aqueous solution and semisolid dosage forms (cream). The in vitro permeation study carried out by using rat skin showed higher permeation, using dendrimers, as compared to aqueous solution of the resveratrol. The advantage of using a PAMAM dendrimer compared to the liposomal preparation is that they are devoid of organic solvents and oils that can be irritating or toxic to the skin [92]. Carletto et al. [93] formulated polycaprolactone nano-capsules loaded with resveratrol (mean particle size 150 nm, PDI < 0.2, and encapsulation efficiency >80%) which are efficient in amorphization of resveratrol and thus improved solubility. With improved solubility, nano-resveratrol formulation significantly increased cytotoxicity in B16F10 melanoma cells compared to the resveratrol solution in in vitro study. In a mouse model bearing B16F10 melanoma tumors following 10 days intraperitoneal treatment (dose 5 mg/kg), the formulation showed decreased tumor volume (2807 mm3 in nanocapsule treated, 9656 mm3 in control and 7940 mm3 in resveratrol solution treated), increased necrotic area and inflammatory infiltrate of melanoma and thus prevented metastasis and pulmonary hemorrhage compared to the free resveratrol [93]. Palliyage et.al. [94] used the combination of curcumin with resveratrol and formulated solid lipid nanoparticles (diameter 180.2 ± 7.7 nm). The combination approach showed in vitro potential to stop metastasis in metastatic B16F10 cell lines based on electrical cell-substrate impedance sensing assay. Further the combination showed synergistic effect in inhibiting the growth of SK-MEL-28 cells [94]. Overall, formulations such as nano-emulsion, liposomes, dendrimers, nano-capsules, etc., have been proven to enhance the solubility and thus permeability of resveratrol across the skin.
References
- Prausnitz, M.R.; Mitragotri, S.; Langer, R. Current status and future potential of transdermal drug delivery. Nat. Rev. Drug Discov. 2004, 3, 115–124.
- Naik, A.; Kalia, Y.N.; Guy, R.H. Transdermal drug delivery: Overcoming the skin’s barrier function. Pharm. Sci. Technol. Today 2000, 3, 318–326.
- Langer, R. Drug delivery and targeting. Nature 1998, 392 (Suppl. 6679), 5–10.
- Bos, J.D.; Meinardi, M.M. The 500 Dalton rule for the skin penetration of chemical compounds and drugs. Experimental Dermatology: Viewpoint 2000, 9, 165–169.
- Wiedersberg, S.; Guy, R.H. Transdermal drug delivery: 30+ years of war and still fighting! J. Control. Release 2014, 190, 150–156.
- Trommer, H.; Neubert, R. Overcoming the stratum corneum: The modulation of skin penetration. Ski. Pharmacol. Physiol. 2006, 19, 106–121.
- Elias, P.M.; Friend, D.S. The permeability barrier in mammalian epidermis. J. Cell Biol. 1975, 65, 180–191.
- Wertz, P.W.; Swartzendruber, D.C.; Squier, C.A. Regional variation in the structure and permeability of oral mucosa and skin. Adv. Drug Deliv. Rev. 1993, 12, 1–12.
- Iyer, R.; Mok, S.; Savkovic, S.; Turner, L.; Fraser, G.; Desai, R.; Jayadev, V.; Conway, A.; Handelsman, D. Pharmacokinetics of testosterone cream applied to scrotal skin. Andrology 2017, 5, 725–731.
- Basaria, S.; Dobs, A.S. New modalities of transdermal testosterone replacement. Treat. Endocrinol. 2003, 2, 1–9.
- Ibrahim, S.A. Spray-on transdermal drug delivery systems. Expert Opin. Drug Deliv. 2015, 12, 195–205.
- Hadgraft, J.; Lane, M.E. Skin permeation: The years of enlightenment. Int. J. Pharm. 2005, 305, 2–12.
- Baroli, B. Penetration of nanoparticles and nanomaterials in the skin: Fiction or reality? J. Pharm. Sci. 2010, 99, 21–50.
- Castro, G.A.; Oréfice, R.L.; Vilela, J.M.; Andrade, M.S.; Ferreira, L.A. Development of a new solid lipid nanoparticle formulation containing retinoic acid for topical treatment of acne. J. Microencapsul. 2007, 24, 395–407.
- Chourasia, R.; Jain, S.K. Drug targeting through pilosebaceous route. Curr. Drug Targets 2009, 10, 950–967.
- Toll, R.; Jacobi, U.; Richter, H.; Lademann, J.; Schaefer, H.; Blume-Peytavi, U. Penetration profile of microspheres in follicular targeting of terminal hair follicles. J. Investig. Dermatol. 2004, 123, 168–176.
- Roberts, M.S. Solute-vehicle-skin interactions in percutaneous absorption: The principles and the people. Ski. Pharmacol. Physiol. 2013, 26, 356–370.
- Chu, K.A.; Yalkowsky, S.H. An interesting relationship between drug absorption and melting point. Int. J. Pharm. 2009, 373, 24–40.
- Yang, R.; Wei, T.; Goldberg, H.; Wang, W.; Cullion, K.; Kohane, D.S. Getting drugs across biological barriers. Adv. Mater. 2017, 29, 1606596.
- Pastore, M.N.; Kalia, Y.N.; Horstmann, M.; Roberts, M.S. Transdermal patches: History, development and pharmacology. Br. J. Pharmacol. 2015, 172, 2179–2209.
- Clemons, M.; Danson, S.; Howell, A. Tamoxifen (‘Nolvadex’): A review: Antitumour treatment. Cancer Treat. Rev. 2002, 28, 165–180.
- Jordan, V.C. Tamoxifen (ICI46, 474) as a targeted therapy to treat and prevent breast cancer. Br. J. Pharmacol. 2006, 147, S269–S276.
- Cuzick, J.; Sestak, I.; Cawthorn, S.; Hamed, H.; Holli, K.; Howell, A.; Forbes, J.F.; Investigators, I.-I. Tamoxifen for prevention of breast cancer: Extended long-term follow-up of the IBIS-I breast cancer prevention trial. Lancet Oncol. 2015, 16, 67–75.
- Waters, E.A.; Cronin, K.A.; Graubard, B.I.; Han, P.K.; Freedman, A.N. Prevalence of tamoxifen use for breast cancer chemoprevention among US women. Cancer Epidemiol. Prev. Biomark. 2010, 19, 443–446.
- Brauch, H.; Schroth, W.; Goetz, M.P.; Mürdter, T.E.; Winter, S.; Ingle, J.N.; Schwab, M.; Eichelbaum, M. Tamoxifen use in postmenopausal breast cancer: CYP2D6 matters. J. Clin. Oncol. 2013, 31, 176.
- Teft, W.A.; Gong, I.Y.; Dingle, B.; Potvin, K.; Younus, J.; Vandenberg, T.A.; Brackstone, M.; Perera, F.E.; Choi, Y.-H.; Zou, G. CYP3A4 and seasonal variation in vitamin D status in addition to CYP2D6 contribute to therapeutic endoxifen level during tamoxifen therapy. Breast Cancer Res. Treat. 2013, 139, 95–105.
- Lee, O.; Page, K.; Ivancic, D.; Helenowski, I.; Parini, V.; Sullivan, M.E.; Margenthaler, J.A.; Chatterton, R.T.; Jovanovic, B.; Dunn, B.K. A randomized phase II presurgical trial of transdermal 4-hydroxytamoxifen gel versus oral tamoxifen in women with ductal carcinoma in situ of the breast. Clin. Cancer Res. 2014, 20, 3672–3682.
- Helland, T.; Hagen, K.B.; Haugstøyl, M.E.; Kvaløy, J.T.; Lunde, S.; Lode, K.; Lind, R.A.; Gripsrud, B.H.; Jonsdottir, K.; Gjerde, J. Drug monitoring of tamoxifen metabolites predicts vaginal dryness and verifies a low discontinuation rate from the Norwegian Prescription Database. Breast Cancer Res. Treat. 2019, 177, 185–195.
- Fontein, D.; Seynaeve, C.; Hadji, P.; Hille, E.; van de Water, W.; Putter, H.; Kranenbarg, E.; Hasenburg, A.; Paridaens, R.J.; Vannetzel, J.-M. Specific adverse events predict survival benefit in patients treated with tamoxifen or aromatase inhibitors: An international tamoxifen exemestane adjuvant multinational trial analysis. J. Clin. Oncol. 2013, 31, 2257–2264.
- Pathan, I.B.; Setty, C.M. Enhancement of transdermal delivery of tamoxifen citrate using nanoemulsion vehicle. Int. J. Pharm. Tech. Res. 2011, 3, 287–297.
- Lin, S.L.; Chan, W.P.; Choy, C.-S.; Leung, T.-K. Enhancement of transdermal delivery of indomethacin and tamoxifen by far-infrared ray-emitting ceramic material (Bioceramic): A pilot study. Transl. Med. 2013.
- Lee, O.; Ivancic, D.; Chatterton, R.T., Jr.; Rademaker, A.W.; Khan, S.A. In vitro human skin permeation of endoxifen: Potential for local transdermal therapy for primary prevention and carcinoma in situ of the breast. Breast Cancer: Targets Ther. 2011, 3, 61.
- Yang, Y.; Pearson, R.M.; Lee, O.; Lee, C.W.; Chatterton, R.T., Jr.; Khan, S.A.; Hong, S. Dendron-Based Micelles for Topical Delivery of Endoxifen: A Potential Chemo-Preventive Medicine for Breast Cancer. Adv. Funct. Mater. 2014, 24, 2442–2449.
- Ambekar, R.S.; Choudhary, M.; Kandasubramanian, B. Recent advances in dendrimer-based nanoplatform for cancer treatment: A review. Eur. Polym. J. 2020, 126, 109546.
- Lin, Y.-L.; Chen, C.-H.; Wu, H.-Y.; Tsai, N.-M.; Jian, T.-Y.; Chang, Y.-C.; Lin, C.-H.; Wu, C.-H.; Hsu, F.-T.; Leung, T.K. Inhibition of breast cancer with transdermal tamoxifen-encapsulated lipoplex. J. Nanobiotechnol. 2016, 14, 1–10.
- Bhatnagar, A.S. The discovery and mechanism of action of letrozole. Breast Cancer Res. Treat. 2007, 105, 7–17.
- Lønning, P.; Dowsett, M.; Powles, T. Postmenopausal estrogen synthesis and metabolism: Alterations caused by aromatase inhibitors used for the treatment of breast cancer. J. Steroid Biochem. 1990, 35, 355–366.
- Perez, E.A. The balance between risks and benefits: Long-term use of aromatase inhibitors. Eur. J. Cancer Suppl. 2006, 4, 16–25.
- Chetrite, G.; Cortes-Prieto, J.; Philippe, J.; Wright, F.; Pasqualini, J. Comparison of estrogen concentrations, estrone sulfatase and aromatase activities in normal, and in cancerous, human breast tissues. J. Steroid Biochem. Mol. Biol. 2000, 72, 23–27.
- Li, L.; Xu, X.; Fang, L.; Liu, Y.; Sun, Y.; Wang, M.; Zhao, N.; He, Z. The transdermal patches for site-specific delivery of letrozole: A new option for breast cancer therapy. AAPS PharmSciTech 2010, 11, 1054–1057.
- Li, L.; Fang, L.; Xu, X.; Liu, Y.; Sun, Y.; He, Z. Formulation and biopharmaceutical evaluation of a transdermal patch containing letrozole. Biopharm. Drug Dispos. 2010, 31, 138–149.
- Maniyar, M.; Chakraborty, A.; Kokare, C. Formulation and evaluation of letrozole-loaded spray dried liposomes with PEs for topical application. J. Liposome Res. 2020, 30, 274–284.
- Geisler, J. Differences between the non-steroidal aromatase inhibitors anastrozole and letrozole–of clinical importance? Br. J. Cancer 2011, 104, 1059–1066.
- Xi, H.; Yang, Y.; Zhao, D.; Fang, L.; Sun, L.; Mu, L.; Liu, J.; Zhao, N.; Zhao, Y.; Zheng, N. Transdermal patches for site-specific delivery of anastrozole: In vitro and local tissue disposition evaluation. Int. J. Pharm. 2010, 391, 73–78.
- Regenthal, R.; Voskanian, M.; Baumann, F.; Teichert, J.; Brätter, C.; Aigner, A.; Abraham, G. Pharmacokinetic evaluation of a transdermal anastrozole-in-adhesive formulation. Drug Des. Dev. Ther. 2018, 12, 3653.
- Mendes, G.D.; Hamamoto, D.; Ilha, J.; dos Santos Pereira, A.; De Nucci, G. Anastrozole quantification in human plasma by high-performance liquid chromatography coupled to photospray tandem mass spectrometry applied to pharmacokinetic studies. J. Chromatogr. B 2007, 850, 553–559.
- An, X.; Tiwari, A.K.; Sun, Y.; Ding, P.-R.; Ashby, C.R., Jr.; Chen, Z.-S. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: A review. Leuk. Res. 2010, 34, 1255–1268.
- Waller, C.F. Imatinib mesylate. In Small Molecules in Hematology; Springer: Cham, Switzerland, 2018; pp. 1–27.
- Das, M.; Shim, K.H.; An, S.S.A.; Yi, D.K. Review on gold nanoparticles and their applications. Toxicol. Environ. Health Sci. 2011, 3, 193–205.
- Larese Filon, F.; Crosera, M.; Adami, G.; Bovenzi, M.; Rossi, F.; Maina, G. Human skin penetration of gold nanoparticles through intact and damaged skin. Nanotoxicology 2011, 5, 493–501.
- Filon, F.L.; Crosera, M.; Timeus, E.; Adami, G.; Bovenzi, M.; Ponti, J.; Maina, G. Human skin penetration of cobalt nanoparticles through intact and damaged skin. Toxicol. In Vitro 2013, 27, 121–127.
- Labala, S.; Mandapalli, P.K.; Kurumaddali, A.; Venuganti, V.V.K. Layer-by-layer polymer coated gold nanoparticles for topical delivery of imatinib mesylate to treat melanoma. Mol. Pharm. 2015, 12, 878–888.
- Moghimi, S.M.; Symonds, P.; Murray, J.C.; Hunter, A.C.; Debska, G.; Szewczyk, A. A two-stage poly (ethylenimine)-mediated cytotoxicity: Implications for gene transfer/therapy. Mol. Ther. 2005, 11, 990–995.
- Labala, S.; Jose, A.; Chawla, S.R.; Khan, M.S.; Bhatnagar, S.; Kulkarni, O.P.; Venuganti, V.V.K. Effective melanoma cancer suppression by iontophoretic co-delivery of STAT3 siRNA and imatinib using gold nanoparticles. Int. J. Pharm. 2017, 525, 407–417.
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: The first drug approved for BRAF-mutant cancer. Nat. Rev. Drug Discov. 2012, 11, 873–886.
- Wellbrock, C.; Hurlstone, A. BRAF as therapeutic target in melanoma. Biochem. Pharmacol. 2010, 80, 561–567.
- da Rocha Dias, S.; Salmonson, T.; van Zwieten-Boot, B.; Jonsson, B.; Marchetti, S.; Schellens, J.H.; Giuliani, R.; Pignatti, F. The European Medicines Agency review of vemurafenib (Zelboraf®) for the treatment of adult patients with BRAF V600 mutation-positive unresectable or metastatic melanoma: Summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur. J. Cancer 2013, 49, 1654–1661.
- Spengler, E.K.; Kleiner, D.E.; Fontana, R.J. Vemurafenib-induced granulomatous hepatitis. Hepatology 2017, 65, 745–748.
- Launay-Vacher, V.; Zimner-Rapuch, S.; Poulalhon, N.; Fraisse, T.; Garrigue, V.; Gosselin, M.; Amet, S.; Janus, N.; Deray, G. Acute renal failure associated with the new BRAF inhibitor vemurafenib: A case series of 8 patients. Cancer 2014, 120, 2158–2163.
- Zou, L.; Ding, W.; Zhang, Y.; Cheng, S.; Li, F.; Ruan, R.; Wei, P.; Qiu, B. Peptide-modified vemurafenib-loaded liposomes for targeted inhibition of melanoma via the skin. Biomaterials 2018, 182, 1–12.
- Ruan, R.; Jin, P.; Zhang, L.; Wang, C.; Chen, C.; Ding, W.; Wen, L. Peptide-chaperone-directed transdermal protein delivery requires energy. Mol. Pharm. 2014, 11, 4015–4022.
- Leman, J.; Dick, D.; Morton, C. Topical 5-ALA photodynamic therapy for the treatment of cutaneous T-cell lymphoma. Clin. Exp. Dermatol. 2002, 27, 516–518.
- Morton, C.; Szeimies, R.M.; Sidoroff, A.; Braathen, L. European guidelines for topical photodynamic therapy part 1: Treatment delivery and current indications–actinic keratoses, Bowen’s disease, basal cell carcinoma. J. Eur. Acad. Dermatol. Venereol. 2013, 27, 536–544.
- Lopez, R.F.V.; Lange, N.; Guy, R.; Bentley, M.V.L.B. Photodynamic therapy of skin cancer: Controlled drug delivery of 5-ALA and its esters. Adv. Drug Deliv. Rev. 2004, 56, 77–94.
- Chen, H.M.; Liu, C.M.; Yang, H.; Chou, H.Y.; Chiang, C.P.; Kuo, M.Y.P. 5-aminolevulinic acid induce apoptosis via NF-κB/JNK pathway in human oral cancer Ca9–22 cells. J. Oral Pathol. Med. 2011, 40, 483–489.
- Pierre, M.B.R.; Tedesco, A.C.; Marchetti, J.M.; Bentley, M.V.L. Stratum corneum lipids liposomes for the topical delivery of 5-aminolevulinic acid in photodynamic therapy of skin cancer: Preparation and in vitro permeation study. BMC Dermatol. 2001, 1, 1–6.
- Lin, M.-W.; Huang, Y.-B.; Chen, C.-L.; Wu, P.-C.; Chou, C.-Y.; Wu, P.-C.; Hung, S.-Y. A formulation study of 5-aminolevulinic encapsulated in DPPC liposomes in melanoma treatment. Int. J. Med. Sci. 2016, 13, 483.
- Touitou, E.; Dayan, N.; Bergelson, L.; Godin, B.; Eliaz, M. Ethosomes—novel vesicular carriers for enhanced delivery: Characterization and skin penetration properties. J. Control. Release 2000, 65, 403–418.
- Allegra, A.; Innao, V.; Russo, S.; Gerace, D.; Alonci, A.; Musolino, C. Anticancer activity of curcumin and its analogues: Preclinical and clinical studies. Cancer Investig. 2017, 35, 1–22.
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Res. 2003, 23, 363–398.
- Liu, W.; Zhai, Y.; Heng, X.; Che, F.Y.; Chen, W.; Sun, D.; Zhai, G. Oral bioavailability of curcumin: Problems and advancements. J. Drug Target. 2016, 24, 694–702.
- Lee, W.-H.; Loo, C.-Y.; Young, P.M.; Traini, D.; Mason, R.S.; Rohanizadeh, R. Recent advances in curcumin nanoformulation for cancer therapy. Expert Opin. Drug Deliv. 2014, 11, 1183–1201.
- Sun, Y.; Du, L.; Liu, Y.; Li, X.; Li, M.; Jin, Y.; Qian, X. Transdermal delivery of the in situ hydrogels of curcumin and its inclusion complexes of hydroxypropyl-β-cyclodextrin for melanoma treatment. Int. J. Pharm. 2014, 469, 31–39.
- Jose, A.; Labala, S.; Venuganti, V.V.K. Co-delivery of curcumin and STAT3 siRNA using deformable cationic liposomes to treat skin cancer. J. Drug Target. 2017, 25, 330–341.
- Kortylewski, M.; Jove, R.; Yu, H. Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 2005, 24, 315–327.
- Yu, H.; Pardoll, D.; Jove, R. STATs in cancer inflammation and immunity: A leading role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809.
- Kanasty, R.; Dorkin, J.R.; Vegas, A.; Anderson, D. Delivery materials for siRNA therapeutics. Nat. Mater. 2013, 12, 967–977.
- Song, X.; Zhang, M.; Dai, E.; Luo, Y. Molecular targets of curcumin in breast cancer. Mol. Med. Rep. 2019, 19, 23–29.
- Atlan, M.; Neman, J. Targeted transdermal delivery of curcumin for breast cancer prevention. Int. J. Environ. Res. Public Health 2019, 16, 4949.
- Abdel-Hafez, S.M.; Hathout, R.M.; Sammour, O.A. Curcumin-loaded ultradeformable nanovesicles as a potential delivery system for breast cancer therapy. Colloids Surf. B Biointerfaces 2018, 167, 63–72.
- Pushpalatha, R.; Selvamuthukumar, S.; Kilimozhi, D. Cyclodextrin nanosponge based hydrogel for the transdermal co-delivery of curcumin and resveratrol: Development, optimization, in vitro and ex vivo evaluation. J. Drug Deliv. Sci. Technol. 2019, 52, 55–64.
- Tian, B.; Liu, J. Resveratrol: A review of plant sources, synthesis, stability, modification and food application. J. Sci. Food Agric. 2020, 100, 1392–1404.
- Carter, L.G.; D’Orazio, J.A.; Pearson, K.J. Resveratrol and cancer: Focus on in vivo evidence. Endocr. Relat. Cancer 2014, 21, R209–R225.
- Walle, T. Bioavailability of resveratrol. Ann. N. Y. Acad. Sci. 2011, 1215, 9–15.
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.; Fong, H.H.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.J.S. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220.
- Athar, M.; Back, J.H.; Tang, X.; Kim, K.H.; Kopelovich, L.; Bickers, D.R.; Kim, A.L. Resveratrol: A review of preclinical studies for human cancer prevention. Toxicol. Appl. Pharmacol. 2007, 224, 274–283.
- Fontecave, M.; Lepoivre, M.; Elleingand, E.; Gerez, C.; Guittet, O. Resveratrol, a remarkable inhibitor of ribonucleotide reductase. FEBS Lett. 1998, 421, 277–279.
- Tsai, M.-J.; Lu, I.-J.; Fu, Y.-S.; Fang, Y.-P.; Huang, Y.-B.; Wu, P.-C. Nanocarriers enhance the transdermal bioavailability of resveratrol: In-vitro and in-vivo study. Colloids Surf. B Biointerfaces 2016, 148, 650–656.
- Hu, C.; Wang, Q.; Ma, C.; Xia, Q. Non-aqueous self-double-emulsifying drug delivery system: A new approach to enhance resveratrol solubility for effective transdermal delivery. Colloids Surf. A Physicochem. Eng. Asp. 2016, 489, 360–369.
- Qi, X.; Wang, L.; Zhu, J.; Hu, Z.; Zhang, J.J.I.j.o.p. Self-double-emulsifying drug delivery system (SDEDDS): A new way for oral delivery of drugs with high solubility and low permeability. Int. J. Pharm. 2011, 409, 245–251.
- Park, S.N.; Jo, N.R.; Jeon, S.H. Chitosan-coated liposomes for enhanced skin permeation of resveratrol. J. Ind. Eng. Chem. 2014, 20, 1481–1485.
- Pentek, T.; Newenhouse, E.; O’Brien, B.; Chauhan, A.S. Development of a topical resveratrol formulation for commercial applications using dendrimer nanotechnology. Molecules 2017, 22, 137.
- Carletto, B.; Berton, J.; Ferreira, T.N.; Dalmolin, L.F.; Paludo, K.S.; Mainardes, R.M.; Farago, P.V.; Favero, G.M. Resveratrol-loaded nanocapsules inhibit murine melanoma tumor growth. Colloids Surf. B Biointerfaces 2016, 144, 65–72.
- Palliyage, G.H.; Hussein, N.; Mimlitz, M.; Weeder, C.; Alnasser, M.H.A.; Singh, S.; Ekpenyong, A.; Tiwari, A.K.; Chauhan, H. Novel Curcumin-Resveratrol Solid Nanoparticles Synergistically Inhibit Proliferation of Melanoma Cells. Pharm. Res. 2021, 38, 851–871.


