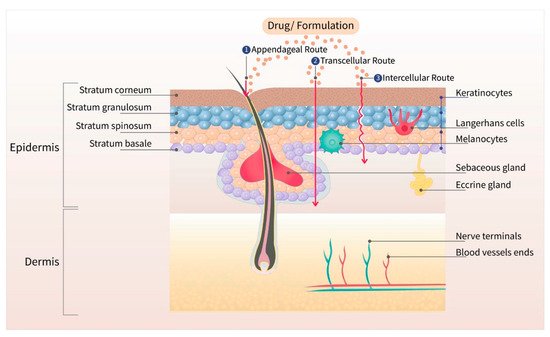
Tamoxifen citrate is available in the form of solution (2 mg/mL as a base), and tablets (10 and 20 mg as a base) for oral administration. It is a trans-isomer of a triphenylethylene derivative, with a molecular weight of 563.6 Da. It acts as a selective estrogen receptor (ER) modulator that competes with β-estradiol for the alpha-estrogen (ERα), thus inhibiting the bioactivity of estrogen in breast tissue [
35,
36]. It is used (1) to treat estrogen receptor-positive (ER
+ve) metastatic breast cancer; (2) as an adjuvant to treat patients with early stage ER
+ve breast cancer; (3) to decrease the risk of invasive breast cancer after tumor excision and radiation in women with ductal carcinoma in situ (DCIS); and (4) to decrease the incidence of breast cancer in women at high risk [
37,
38]. The activity of the drug is due to the metabolites 4-hydroxytamoxifen (4-OHT) and N-desmethyl-4-hydroxytamoxifen (ENX), which are formed by the cytochrome P450 enzymes, CYP2D6 and CYP3A4/5, which have a higher affinity for ERα compared to tamoxifen [
39,
40].
Although the oral formulation significantly decreases the risk of recurrence of ER
+ve DCIS, its use can achieve the following: (1) activate ERα receptors in the endometrium (increasing the risk of cancer); (2) increase the risk of venous thromboembolic events; (3) cause vasomotor symptoms and vaginal symptoms such as dryness, discharges, and atrophy [
41,
42,
43]. Furthermore, the efficacy of oral tamoxifen could be decreased in women with polymorphisms in CYP2D6 and CYP3A4/5 as this would decrease the levels of tamoxifen’s active metabolites [
39,
40]. The topical delivery of 4-OHT gel to the local tumors was shown to be a suitable alternative for oral delivery, decreasing the risk of systemic adverse effects. A randomized, pre-surgical trial in pre- and post-menopausal women was conducted by comparing transdermal 4-OHT gel (4 mg/day) to the oral tamoxifen (20 mg/day) [
41]. The results indicated that equivalent concentrations of 4-OHT, i.e., 5.4 and 5.8 ng/g, were obtained in breast tissue biopsy samples from patients treated with oral and transdermal formulation, respectively. There was no significant correlation between the amount of 4-OHT in the tissue and the plasma in the transdermal formulation treated group. Thus, using the transdermal route sufficient amount of drug can be delivered to the target tissue without spiking the 4-OHT concentration in the blood. In contrast, with the oral tamoxifen group, there was a significant correlation between the concentration of 4-OHT in the plasma and the tissue leading to a spike in drug concentration, thereby causing systemic side effects. Therefore, the transdermal 4-OHT gel was found to be superior when compared to oral tamoxifen in localizing 4-OHT in patients with DCIS, thus decreasing the incidence of adverse systemic side effects, and thereby increasing the patient compliance [
41]. In other study, Pathan et al. optimized a nano-emulsion of tamoxifen citrate by using arachis oil, Cremophore EL, and ethanol [
44]. The solubility of tamoxifen was highest in arachis oil compared to other oils such as jojoba oil, coconut oil, castor oil and sesame oil. Among different surfactants and co-surfactants (Labrafil, Tween-80, Cremopore EL, ethanol, butanol, and propanol), Cremophore EL and ethanol demonstrated a better solubility profile for tamoxifen citrate. The optimized nano-emulsion possessed surfactant to co-surfactant ratio of 1:1, surfactant to oil ratio of 1:9, and a drug content of 5%
w/w. This optimized formulation displayed a mean droplet size of 68 nm with a polydispersity index (PDI) of 0.125, and viscosity of 201 cP. The flux of tamoxifen evaluated by using Keshary–Chien diffusion cells across the excised rat skin was found to be 98.98 µg/cm
2/h [
44]. However, a control formulation of tamoxifen was not used to compare the efficacy of the formulation. Lin et al. [
45] used bioceramic irradiation, with an emissivity of 0.98 at a wavelength of 6 to 14 μm, to enhance the transdermal delivery of tamoxifen. The permeation across cellulose acetate membrane showed that the bioceramic irradiation on water molecules weakens the hydrogen bond, which decreases the viscosity, thereby enhances the permeation. The study demonstrated higher permeation of tamoxifen and indomethacin by using bioceramic irradiation compared to the control, where bioceramics were not used [
45]. Similarly, Lee et al. [
46] evaluated the relative efficiency of the skin permeation of 4-OHT and ENX, using split-thickness human skin. The permeation of ENX across human skin was improved by using oleic acid as a permeation enhancer. However, the permeation of 4-OHT was significantly lower than that of ENX, even in the presence of oleic acid [
46]. Dendrimer-based micelles were prepared by Yang et al. [
47] to determine the feasibility of delivering ENX by the transdermal route. Generally, dendrimer-based drug-delivery systems involve the chemical conjugation of the drug with the surface groups of dendrimers to increase the drug stability during transport [
48]. Due to a limited number of reactive functional groups on ENX, dendrimer conjugation was not feasible. Therefore, PEGylated dendro-based copolymers (PDCs) that can self-assemble into dendron micelles (DM) were utilized for ENX delivery [
47]. The solubility and sequestration of ENX in DM was higher (3% drug loading efficiency, mean size of 48.4 nm), with smaller and uniform particle size distribution compared to the ENX cationic liposomes (0.1% drug loading efficiency, mean size of 100 nm) made with DOTAP, DMPC and cholesterol at a molar ratio of 2:2:1. Furthermore, cell titer 96 aqueous one solution (MTS) assay in ER
+ve MCF-7 and ER
-ve MDA-MB-231 breast cancer cell lines indicated that the ER-dependent, anti-proliferative efficacy of ENX was retained after encapsulation in DMs. In contrast to the liposomal formulation, the permeation of ENX from DMs across rat and human skin were sustained over 6 days. However, the intactness of the skin at the end of the study was not demonstrated. The flux of the ENX across the skin from DMs was proportional to the sequestered drug in the DMs. Despite poor drug loading, the liposomes demonstrated the highest permeability coefficient (K
p 0.0467 cm/h). Moreover, the DMs (size >24 nm) cannot permeate through the aqueous pores of the skin (size <4 nm), as they have a significantly larger size compared to the aqueous pores of the skin. However, DMs facilitate the permeation of drugs through the skin by translocating the drug molecules [
47].
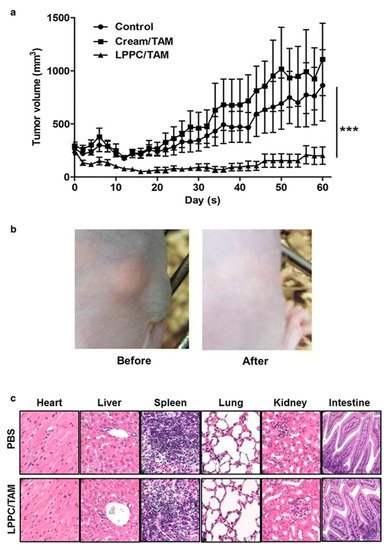
Lin et al. [
49] used the liposome–PEG–PEI complex (LPPC) for the transdermal delivery of tamoxifen in vivo in mice xenograft with BT474 breast cancer cells. The in vitro efficacy of the LPPC-tamoxifen was determined in the breast cancer cell lines, ER
+ve MCF-7, DT474 and ER
-ve MDA-MB-231, using the 3-(4,5-dimethylthiazol-2-yl)-2, 5-diphenyl tetrazolium bromide (MTT assay). Although the IC
50 values were not determined, the liposomal formulation significantly reduced the viability of the ER
+ve and ER
-ve cell lines. Tamoxifen arrested the proliferation of ER
-ve breast cancer cell lines in the S phase of the cell cycle. Furthermore, the study showed that the inhibition of the CIP2A/PP2A/p-Akt (Protein phosphatase 2A (PP2A), a cellular inhibitor of PP2A/CIPP2A, protein kinase B-Akt) signaling pathway was responsible for the inhibition of ER
-ve cell proliferation. The LPPC-tamoxifen (LPPC/TAM) decreased tumor growth by 82% compared to the control. LPPC/TAM was also found to effectively inhibit BT474 tumor growth than cream-tamoxifen. In addition, the tissue damage and pathology of organs induced by LPPC/TAM treatment were assessed. The results in the treated mice showed that LPPC/TAM did not cause any skin irritation or injury to organs (
Figure 2). Overall, the results indicated the feasibility of using LPPC formulations for the local delivery of tamoxifen in breast cancer cells in an in vivo mouse model [
49]. Several research studies demonstrated that local delivery of tamoxifen and its derivatives via transdermal route is possible. Moreover, the compounds disperse in a gel matrix or in the form of nanomedicines (ethosomes, liposomes, dendrimers, etc.) and can provide effective drug levels at the tumor sites in animal models.