 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Saori Furuta | + 16977 word(s) | 16977 | 2021-07-28 08:46:01 | | | |
| 2 | Vivi Li | -6939 word(s) | 10038 | 2021-08-09 12:32:48 | | |
Video Upload Options
Nitric oxide (NO) is a highly reactive molecule, generated through metabolism of L-arginine by NO synthase (NOS). Abnormal NO levels in mammalian cells are associated with multiple human diseases, including cancer. Recent studies have uncovered that the NO signaling is compartmentalized, owing to the localization of NOS and the nature of biochemical reactions of NO, including S-nitrosylation. S-nitrosylation is a selective covalent post-translational modification adding a nitrosyl group to the reactive thiol group of a cysteine to form S-nitrosothiol (SNO), which is a key mechanism in transferring NO-mediated signals. While S-nitrosylation occurs only at select cysteine thiols, such a spatial constraint is partially resolved by transnitrosylation, where the nitrosyl moiety is transferred between two interacting proteins to successively transfer the NO signal to a distant location. As NOS is present in various subcellular locales, a stress could trigger concerted S-nitrosylation and transnitrosylation of a large number of proteins involved in divergent signaling cascades. S-nitrosylation is an emerging paradigm of redox signaling by which cells confer protection against oxidative stress.
1. Introduction
In the past decades, nitric oxide (NO) has garnered an increasing amount of interest with regard to its impact on many human diseases. Since its discovery by Furchgott et al. as “the endothelium-derived relaxing factor (EDRF)” over 30 years ago [1], NO has become one of the most studied subjects in the field of biomedical sciences. NO was named “the Molecule of Year” in 1992, and its discovery led to the 1998 Nobel Prize of Furchgott et al. Despite the original conception of NO as a freely diffusible gas [2], NO production and signaling are rather compartmentalized and spatially regulated. This is owing to the fact that all the isoforms of NO synthases (NOS1-3) are mostly localized at the membrane and organelles [3][4][5], and because NO produced there is quickly (<0.1 sec) consumed by reacting with the proximal molecules [5][6][7]. On one hand, NO reacts with molecular oxygen to become nitrogen oxide, which then reacts with a transition metal or cysteine thiol to form a nitrosyl adduct (metal nitrosylation or S-nitrosylation, respectively). The nitrosyl group could be further transferred to another protein in a distant site (transnitrosylation) to propagate NO signaling. Such effects of NO are said to be “direct” and involved in many anti-oxidant mechanisms as well as other biochemical activities [8]. On the other hand, in the presence of high levels of reactive oxygen species (ROS), such as superoxide, NO reacts with ROS and forms a strong oxidant, reactive nitrogen species, which in turn reacts with and oxidatively damages a variety of biological molecules. This mode of NO effect is said to be “indirect” and involved in “pro-oxidant” signaling of NO [8].
Initial NO studies focused on the NO signaling pathway in specialized tissues such as neurons, muscles, endothelia and immune cells. For example, in the brain NO controls brain-blood flow and other processes that lead to normal brain function. In the circulatory system, NO controls leucocyte adhesion, pro-inflammatory signaling, platelet aggregation, and angiogenesis [9]. Such diverse functions of NO are mediated by the “classical” and “non-classical” schemes. In the classical NO signaling, NO binds the heme iron of guanylyl cyclase (sGC) to induce production of cGMP, which then activates the cGMP-dependent protein kinase (PKG) pathway [10]. Notwithstanding this classical scheme, recent studies have unveiled a wealth of “non-classical” NO signaling that mediates pleiotropic functions in diverse tissues/organs. A major mechanism of non-classical NO’s functions is S-nitrosylation, an NO-dependent covalent modification of a cysteine thiol [11]. S-nitrosylation triggers a change in the protein structure, which could alter protein-protein interactions and enable further post-translational modifications such as phosphorylation, acetylation, ubiquitination, and disulfide bond formation [11][12][13][14]. Furthermore, in the presence of a low level of ROS, S-nitrosylation could not only scavenge NO to prevent it from reacting with ROS, but also protect cysteine thiols against ROS-mediated oxidation [sulfonic acid (RSO3H) formation] [15]. At very high levels of ROS, however, NO could react with ROS to form reactive nitrogen species as discussed above [8][16].
S-nitrosylation could regulate >3000 proteins involved in a multitude of biological processes, including protein stability/turnover, steroid synthesis, transcription regulation, DNA damage repair, cellular growth/differentiation, apoptosis and redox regulation. Thanks to various subcellular locales of NOS, a stress could induce concerted S-nitrosylation and transnitrosylation of proteins involved in divergent signaling cascades. Dysregulated S-nitrosylation has been implicated in the incidence and progression of different diseases [11][15][17][18][19]. In this review, we will provide the recent advances in our understanding of how S-nitrosylation exerts redox regulation on cells and tissues. We will especially focus on cellular locales which are equipped with various mechanisms that produce or fight against ROS, such as the mitochondrion, nucleus, and extracellular environment. We will then discuss how dysregulated S-nitrosylation would lead to disease conditions.
2. NO and Its Roles in Redox Regulation
NO is a reactive, gaseous signaling molecule with a half-life of 0.09–2 seconds [6]. NO is produced by nitric oxide synthase (NOS) using amino acid L-arginine as the substrate in a wide range of tissues and is involved in pleiotropic functions. Most NO studies have focused on its role in specialized cells, including neurons, muscles, endothelia, and immune cells. In these cells, NO mediates neurotransmission, muscle and vessel dilation, and pro-inflammatory signaling. Aside from these well-established roles, NO exerts pleiotropic functions in many different types of cells [20]. These functions include regulation of tissue morphogenesis, polarity formation, cellular growth, and movement [21][22][23][24][25][26].
Because of its nature as a radical, NO (•NO) is also involved in both anti- and pro-oxidant mechanisms. As an anti-oxidant, NO could modify cellular processes to confer protection to cells and tissues against oxidative damage [8]. It is reported that pretreatment with NO could protect cells against the oxidative stress by hydrogen peroxide (H2O2) [27][28], while NO could also limit the production of ROS by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in immune cells [29]. In addition, the beneficial effects of pretreatment with NO against ischemia–reperfusion-mediated tissue injury have been reported in clinical trials [30]. Conversely, as a pro-oxidant, NO could react with molecular oxygen or ROS (especially, superoxide: O2•−) and be converted to a strong oxidant, reactive nitrogen oxide species (RNOS, especially, peroxynitrite: ONOO−), that could oxidatively damage a variety of biological molecules.
It is proposed that whether NO exerts anti- or pro-oxidant effects depends on the concentration of NO [8]. At lower NO concentrations, the activities of NO manifest through its direct interaction with the biological target, which are likely to lead to anti-oxidant effects. At higher NO concentrations, conversely, the activities of NO are indirectly mediated by RNOS derived from the reaction of NO with molecular oxygen or ROS, which are likely to lead to pro-oxidant effects. The deleterious effects of NO as a pro-oxidant often take place in confined locales where local NO concentrations could be quickly raised to higher levels [8]. In fact, dose-dependent activities of NO have been reported by a number of previous studies. These studies suggest that the threshold concentration of NO that determines whether the effect is direct or indirect would be around 1 μM [8][31].
Local NO concentration is in part controlled by spatial regulation and compartmentalization of the NO source. There are three isoforms of NOS (NOS1-3) that produce NO in mammalian cells. NOS-1 (neuronal NOS, nNOS) and NOS-3 (endothelial NOS, eNOS) are the constitutive NOS and are part of the membrane-bound protein complexes found in different subcellular locales. NO produced by NOS-1 or NOS-3 quickly (<0.1 sec.) and directly reacts with the proximal targets, including NOS itself and the interacting proteins [3][4][6]. In contrast, NOS-2 (iNOS) is the inducible NOS. In inflammatory cells, NOS-2 is initially expressed in the cytosol, but is recruited to phagosomes or peroxisomes to elevate the local NO concentration, where NO reacts with superoxide, forming peroxynitrite to kill pathogens [32][33]. Thus, dichotomous effects of NO as both an anti- and pro-oxidant are dependent on the source and location of NO that determines its local concentration.
3. NO Production and Biochemistry
3.1. Nitric Oxide Synthase (NOS)
Mammalian NOS consists of three isoforms (NOS1-3) that encompass ~50% homology [7]. NOS-1 (nNOS) and-3(eNOS) are expressed constitutively, while their activities are regulated post-translationally, such as by phosphorylation, S-nitrosylation, protein interaction, cofactor/substrate, and calcium level. NOS-1 and NOS-3 produce a steady-state NO level to regulate tissue homeostasis and development [10][34]. Conversely, the expression of NOS-2 (iNOS) is regulated inducibly to produce a large amount of NO in response to an inflammatory signal [10][34]. In addition, mitochondria are reported to possess mtNOS (nNOS homologue) in the matrix and inner membrane. mtNOS is involved in the regulation of oxygen consumption and biogenesis of mitochondria [35][36][37][38][39]. However, the actual presence of mtNOS is currently under debate (see below for details).
NOS is a dimer of two identical monomers tethered by tetrahedral coordination of a zinc ion at two CysXXXXCys motifs (each motif is contributed by a monomer). Through the motif, NOS binds the substrate L-arginine and cofactor tetrahydrobiopterin (BH4) which facilitates dimerization, substrate binding, and enzymatic function [34][40]. The constitutive NOS-1 and -3, but not inducible NOS-2, are bound by calmodulin in response to increased calcium levels, which induces conformational changes to activate the enzymatic function [34][41]. Each NOS monomer is composed of two distinct domains: the carboxyl-terminal reductase and amino-terminal oxygenase domains. Both domains are connected by a linker region that binds calmodulin [34][41]. The reductase domain harbors a series of redox-active cofactors, NADPH, flavin adenine dinucleotide (FAD), and flavin mononucleotide (FMN), which serially transfers electrons. The oxygenase domain, serving as the dimerization site that coordinates a zinc ion, harbors another redox-active cofactor BH4, heme, and the substrate L-arginine. In particular, BH4 plays an essential role in “coupling” the reductase and oxygenase domains [7][34][42][43]. It facilitates electron transfer from FMN of the reductase domain to the heme of the oxygenase domain. This causes the ferric (Fe3+) center of the heme to be reduced to the ferrous (Fe2+) center, which then reacts with molecular oxygen to form Fe2+-O2 complex. This complex, in turn, oxidizes the guanidine moiety of the substrate L-arginine, producing L-citrulline and NO [7][34][42][43] (Figure 1, left).
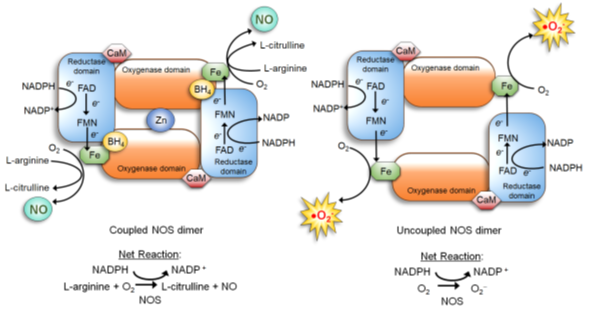
Figure 1. Nitric Oxide Synthase (NOS) dimer in the coupled (normal) vs. uncoupled states [40]. (Left) In the normal coupled state, two NOS monomers are tethered by BH4, which also increases the binding affinity of the substrate L-arginine. Furthermore, the NOS dimer is stabilized by a zinc ion coordinated in the oxygenase domains of two monomers. Dimerization allows the coupling of two reactions: 1) electron flow: nicotinamide adenine dinucleotide phosphate (NADPH)→ flavin adenine dinucleotide (FAD)→ flavin mononucleotide (FMN)→ Fe of the heme→ molecular oxygen; and 2) oxidation of L-arginine. This coupling yields NO and L-citrulline. (Right) When the BH4 level is scarce, NOS remains as a monomer and fails to bind L-arginine. The electron flow to molecular oxygen (Reaction 1) is uncoupled to L-arginine oxidation (Reaction 2), yielding superoxide (O2•−).
3.2. NO Signaling
3.3. S-Nitrosylation, Metal Nitrosylation and Nitration
4. S-Nitrosylation
4.1. Biochemistry of S-Nitrosylation
NO itself is not an oxidant and does not strongly react with a protein thiol. Thus, for the majority of S-nitrosylation, NO first reacts with oxygen to increase the oxidation state and then reacts with a thiol [73]. Different intermediate reactions have been proposed [56].
1) First, NO reacts with O2 to form a series of nitrogen oxides with increasing oxidation states (auto-oxidation). N2O3 reacts with a protein thiol to produce nitrite and a nitrosothiol. In particular, the rate of this reaction increases in hydrophobic environments such as membranes, the predominant locales of NOS1-3 (Figure 2, reaction 1) [56].
2) First, NO reacts with O2 to form NO2. NO2 reacts with a thiol to produce a thiol radical and nitrite. Then, NO reacts with a thiol radical to form a nitrosothiol (Figure 2, reaction 2) [56].
3) In the presence of a thiol radical, NO directly reacts with it to form a nitrosothiol (Figure 2, reaction 3) [56].
4) For a metal-catalyzed reaction, NO becomes oxidized by a transition metal, such as Fe3+ or Cu2+, in a metalloenzyme, forming nitrosonium (NO+). Nitrosonium then reacts with a thiol close to the catalytic center to form a nitrosothiol. This reaction takes place in specific cases, such as auto-nitrosylation of hemoglobin and GSNO formation by cytochrome c (Figure 2, reaction 4) [56].
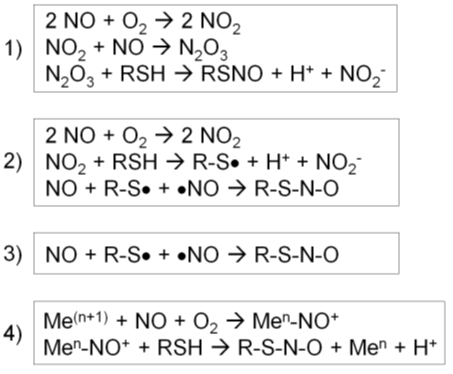
Figure 2. Four different types of S-nitrosylation reactions [56]. (1) Nitric oxide (NO) reacts with O2 to form a series of nitrogen oxides. N2O3 reacts with a protein thiol to produce nitrite and a nitrosothiol. (2) NO reacts with O2 to form NO2, which reacts with a thiol to produce a thiol radical and nitrite. Then, NO reacts with a thiol radical to form a nitrosothiol. (3) In the presence of a thiol radical, NO directly reacts with it to form a nitrosothiol. (4) NO becomes oxidized by a transition metal, forming nitrosonium (NO+). Nitrosonium then reacts with a thiol close to the catalytic center to form a nitrosothiol.
4.2. S-Nitrosylation Reaction Specificity
4.3. Other Factors that Control S-Nitrosylation Level
5. Transnitrosylation
5.1. Reaction
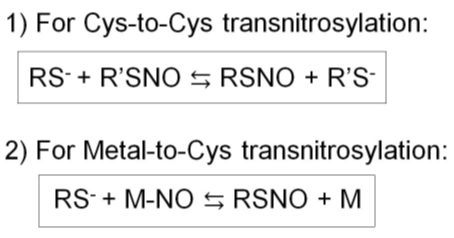
Figure 3. Two types of transnitrosylation reactions. 1) Cys-to-Cys transnitrosylation. 2) Metal-to-Cys transnitrosylation.
5.2. S-Nitrosylases or Transnitrosylases
5.2.1. S-Nitrosoglutathione (GSNO)
5.2.2. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
5.2.3. S100A8/S100A9
6. S-Nitrosylation of Cellular Proteins for Redox Regulation
Inside the cell, S-nitrosylation levels of cytosolic proteins are relatively low. One reason for this is that the S-nitrosyl group could be spontaneously cleaved by various factors in the cytosol. These factors include reducing agents (ascorbate and glutathione), metal ions (Cu2+), heat, UV, ROS, and nucleophiles (or anions) [17][91]. The other reason is that the subcellular localizations of the constitutive NOS-1 and NOS-2 are compartmentalized, which also compartmentalizes the majority of S-nitrosylation reactions [3][4][6][117]. Although NOS-2 is initially expressed in the cytosol, NO produced by NOS-2 is more likely to react with ROS to form RNOS than S-nitrosylating protein thiols [32][33]. Thus, the major sites of S-nitrosylation are organelles, (e.g., mitochondria, Golgi apparatus, and endoplasmic reticulum), nucleus, and plasma membrane. Among these sites, we will feature mitochondria and nuclei where S-nitrosylation of proteins plays critical roles in protection of cells against oxidative stress. Some of these proteins are listed in Table 1.
| Protein Name | Effect of S-Nitrosylation | Consequence | Ref. |
|---|---|---|---|
| Bcl-2 | Inhibition of ubiquitination (activation) | Anti-apoptosis | [18] |
| Caspases | Inhibition of caspase activity | Anti-apoptosis | [19][119] |
| Fas Receptor | Promote Fas ligand mediated apoptosis | Apoptosis | [18] |
| GAPDH | Nuclear translocation | Apoptosis | [105] |
| TG2 | Inhibition of activity | Tumor suppression | [120][121] |
| MGMT | Inhibition of DNA repair activity | DNA damage & apoptosis | [18][122][123] |
| OGG1 | Inhibition of DNA repair activity | DNA damage & apoptosis | [122] |
| HDAC2 | Inhibition of deacetylase activity | Histone acetylation | [124][125] |
| MMPs | Activation (low level) Inhibition (high level) |
Tumor progression Tumor suppression |
[126][127] |
| Caveolin-1 | Inhibition of proteasomal degradation | Tumor progression | [128] |
| c-Src | Activation | Tumor progression | [18] |
| Β-catenin | Proteasomal degradation | Tumor suppression | [129] |
| HDM2 | Inhibition of binding to p53 | Tumor suppression | [130] |
| HIF-1α | Stabilization of activity | Pro-angiogenesis | [14][18] |
| MAT | Inactivation of enzyme activity | Tumor suppression | [82][131] |
| NF-kB | Inhibition of activity | Tumor suppression | [14] |
| PTEN | Inhibition of activity | Tumor progression | [18][132] |
| Ras | Activation | Tumor progression | [133] |
6.1. S-Nitrosylation of Redox-Sensitive Mitochondrial Proteins
Mitochondria play key roles in the production of ROS as well as conferring antioxidant mechanisms along with all other functions (Figure 4) [134]. The bioactivities and quality control of mitochondria are in part regulated by S-nitrosylation and denitrosylation of mitochondrial proteins [135][136]. Mitochondrial proteins become S-nitrosylated in response to changes in mitochondrial respiration and redox equilibrium. This results in the protection of protein thiols and the cell against oxidative damage and death, while also preventing further ROS production [59][60][61]. Such responsiveness of S-nitrosylation to the oxidizing environment could be in part ascribed to the nature of its biochemistry. NO itself is not an oxidant and would need to react with molecular oxygen to increase the oxidation state in order to react with a protein thiol [73].
The source of mitochondrial NO remains under debate. On one hand, mitochondrial NO may be generated by mitochondrial NOS (mtNOS) [60]. mtNOS is reported to be calcium-sensitive and constitutively active (homologous to NOS-1 [nNOS]) and integral to the inner mitochondrial membrane [36]. However, the existence of mtNOS remains controversial [60]. On the other hand, mitochondrial NO may be derived from NO generated in the cytoplasm and transported via a transnitrosylase through a cognate transporter on the mitochondrial membranes [61][137].
NO produced in the mitochondria could S-nitrosylate or Fe-nitrosylate proteins preferentially in the inner mitochondrial membrane and intermembrane space because of their lower pH and lipophilic environments. Mitochondrial proteins targeted for S-nitrosylation include all the complexes in the electron transport chain (ETC): Complex I (NADH: ubiquinone oxidoreductase); Complex II (succinate dehydrogenase); Complex III (cytochrome b-c1 Complex); cytochrome c, Complex IV (cytochrome C oxidase) as well as adenosine triphosphate (ATP) synthase (Complex V) [59][60]. In addition, several metabolic enzymes involved in citric acid (Krebs) cycle and β-oxidation [60][138], as well as different mitochondrial proteins involved in regulation of apoptosis [119][139][140], are S-nitrosylated. Such S-nitrosylation of mitochondrial proteins mostly inhibit their activities to regulate mitochondrial O2 consumption, ROS production, and mitophagy, while protecting the cell against death signals (Figure 4) [36].
On the other hand, in the presence of large amounts of ROS (and/or NO), NO could react with superoxide to produce a powerful oxidant, peroxynitrite (ONOO−) that can irreversibly nitrate or peroxidate tyrosines and unsaturated fatty acids [137][141].
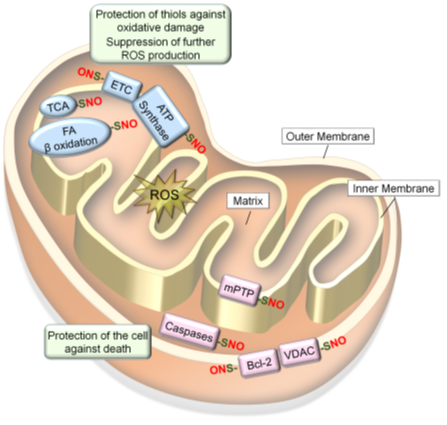
Figure 4. S-nitrosylation of mitochondrial proteins confer resistance to oxidative stress. Mitochondrial proteins become S-nitrosylated in oxidizing environments. S-nitrosylation of proteins involved in respiration and energy production (ETC, ATP synthesis, TCA cycle and fatty acid β oxidation) leads to prevention of ROS production and protection of protein thiols in these enzymes against oxidative damage. In contrast, S-nitrosylation of proteins involved in cell death regulation (mPTP, caspases-3/-9, VDAC and Bcl-2) leads to protection of the cell against death signals.
6.1.1. Electron Transport Chain (ETC) and ATP Synthase
6.1.2. Metabolic Enzymes
6.1.3. Mitochondrial Pro- and Anti-Apoptotic proteins
6.2. S-Nitrosylation of REDOX-Regulatory Nuclear Proteins
6.2.1. Transcription Factors
6.2.2. Nuclear Translocator
6.2.3. DNA Damage Repair Proteins
7. S-Nitrosylation of Extracellular Proteins for Redox Regulators
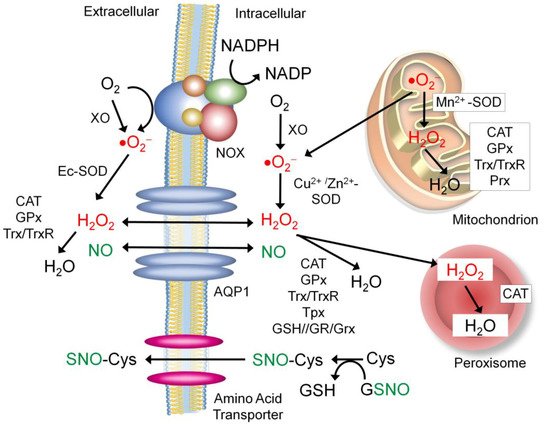
7.1. Sources of the Extracellular NO and Nitrosyl Group
7.2. Proteins that Regulate Extracellular ROS and NO Levels
7.2.1. Aquaporin-1 (APQ-1)
7.2.2. NADPH Oxidase (NOX)
7.2.3. Xanthine Oxidase (XO)
7.2.4. Extracellular Superoxide Dismutase (Ec-SOD)
7.3. Other Secretory Proteins Regulated by S-nitrosylation for Redox Control
7.3.1. Antioxidant Enzymes
7.3.2. Protein Disulfide Isomerase (PDI)
8. Dysregulated S-Nitrosylation in Disease Pathogenesis
8.1. Tumor Microenvironment (TME) and Oxidative Stress
8.2. Matrix Metalloproteinases (MMPs)
8.3. Tissue Transglutaminase (TG2)
9. Conclusions
References
- Furchgott, R.F.; Cherry, P.D.; Zawardzki, J.V.; Jothianandan, D. Endothelial cells as mediators of vasodilation of arteries. J. Cardiovasc. Pharmacol. 1984, 6, S336–S343.
- Cudeiro, J.; Rivadulla, C.; Rodríguez, R.; Martínez-Conde, S.; Martínez, L.; Grieve, K.L.; Acuña, C. Further observations on the role of nitric oxide in the feline lateral geniculate nucleus. Eur. J. Neurosci. 1996, 8, 144–152.
- Ho, G.P.; Selvakumar, B.; Mukai, J.; Hester, L.D.; Wang, Y.; Gogos, J.A.; Snyder, S.H. S-nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron 2011, 71, 131–141.
- Martínez-Ruiz, A.; Villanueva, L.; González de Orduña, C.; López-Ferrer, D.; Higueras, M.A.; Tarín, C.; Rodríguez-Crespo, I.; Vázquez, J.; Lamas, S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc. Natl. Acad. Sci. USA 2005, 102, 8525–8530.
- Nakamura, T.; Lipton, S.A. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol. Sci. 2016, 37, 73–84.
- Thomas, D.D.; Liu, X.; Kantrow, S.P.; Lancaster, J.R.J. The biological lifetime of nitric oxide: Implications for the perivascular dynamics of NO and O2. Proc. Natl. Acad. Sci. USA 2001, 98, 355–360.
- Alderton, W.K.; Cooper, C.E.; Knowles, R.G. Nitric oxide synthases: Structure, function and inhibition. Biochem. J. 2001, 357 Pt 3, 593–615.
- Wink, D.A.; Miranda, K.M.; Espey, M.G.; Pluta, R.M.; Hewett, S.J.; Colton, C.; Vitek, M.; Feelisch, M.; Grisham, M.B. Mechanisms of the antioxidant effects of nitric oxide. Antioxid. Redox Signal. 2001, 3, 203–213.
- Luiking, Y.C.; Engelen, M.P.; Deutz, N.E. Regulation of nitric oxide production in health and disease. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 97–104.
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563.
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166.
- Hess, D.T.; Stamler, J.S. Regulation by S-nitrosylation of protein post-translational modification. J. Biol. Chem. 2012, 287, 4411–4418.
- Selvakumar, B.; Jenkins, M.A.; Hussain, N.K.; Huganir, R.L.; Traynelis, S.F.; Snyder, S.H. S-nitrosylation of AMPA receptor GluA1 regulates phosphorylation, single-channel conductance, and endocytosis. Proc. Natl. Acad. Sci. USA 2013, 110, 1077–1082.
- Martínez-Ruiz, A.; Lamas, S. S-nitrosylation: A potential new paradigm in signal transduction. Cardiovasc. Res. 2004, 62, 43–52.
- Sun, J.; Steenbergen, C.; Murphy, E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid. Redox Signal. 2006, 8, 1693–1705.
- Jerca, L.; Jerca, O.; Mancas, G.; Constantinescu, I.; Lupuşoru, R. Mechanism of action and biochemical effects of nitric oxide. J. Prev. Med. 2002, 10, 35–45.
- Kovacs, I.; Lindermayr, C. Nitric oxide-based protein modification: Formation and site-specificity of protein S-nitrosylation. Front. Plant Sci. 2013, 4, 137.
- Wang, Z. Protein S-nitrosylation and cancer. Cancer Lett. 2012, 320, 123–129.
- Plenchette, S.; Romagny, S.; Laurens, V.; Bettaieb, A. S-Nitrosylation in TNF superfamily signaling pathway: Implication in cancer. Redox Biol. 2015, 6, 507–515.
- Antosova, M.; Plevkova, J.; Strapkova, A.; Buday, T. Nitric oxide—Important messenger in human body. OJMIP 2012, 2, 98–106.
- Peunova, N.; Scheinker, V.; Ravi, K.; Enikolopov, G. Nitric oxide coordinates cell proliferation and cell movements during early development of Xenopus. Cell Cycle 2007, 6, 3132–3144.
- Young, S.L.; Evans, K.; Eu, J.P. Nitric oxide modulates branching morphogenesis in fetal rat lung explants. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L379–L385.
- Cole, A.G.; Mashkournia, A.; Parries, S.C.; Goldberg, J.I. Regulation of early embryonic behavior by nitric oxide in the pond snail Helisoma trivolvis. J. Exp. Biol. 2002, 205 Pt 20, 3143–3152.
- Slezinger, M.S.; Kuzin, B.A. Nitric oxide synthase mediates regulation of cell polarity and movement during Drosophila melanogaster morphogenesis. Ontogenez 2009, 40, 40–47.
- Bradley, S.; Tossell, K.; Lockley, R.; McDearmid, J.R. Nitric oxide synthase regulates morphogenesis of zebrafish spinal cord motoneurons. J. Neurosci. 2010, 30, 16816–16831.
- Lee, N.P.; Cheng, C.Y. Nitric oxide/nitric oxide synthase, spermatogenesis, and tight junction dynamics. Biol. Reprod. 2004, 70, 267–276.
- Nunoshiba, T.; DeRojas-Walker, T.; Wishnok, J.S.; Tannenbaum, S.R.; Demple, B. Activation by nitric oxide of an oxidative-stress response that defends Escherichia coli against macrophages. Proc. Natl. Acad. Sci. USA 1993, 90, 9993–9997.
- Kim, Y.M.; Bergonia, H.; Lancaster, J.R.J. Nitrogen oxide-induced autoprotection in isolated rat hepatocytes. FEBS Lett. 1995, 374, 228–232.
- Clancy, R.M.; Leszczynska-Piziak, J.; Abramson, S.B. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J. Clin. Investig. 1992, 90, 1116–1121.
- Bice, J.S.; Jones, B.R.; Chamberlain, G.R.; Baxter, G.F. Nitric oxide treatments as adjuncts to reperfusion in acute myocardial infarction: A systematic review of experimental and clinical studies. Basic Res. Cardiol. 2016, 111, 23.
- Thomas, D.D.; Ridnour, L.A.; Isenberg, J.S.; Flores-Santana, W.; Switzer, C.H.; Donzelli, S.; Hussain, P.; Vecoli, C.; Paolocci, N.; Ambs, S.; et al. The chemical biology of nitric oxide: Implications in cellular signaling. Free Radic Biol. Med. 2008, 45, 18–31.
- Loughran, P.A.; Stolz, D.B.; Barrick, S.R.; Wheeler, D.S.; Friedman, P.A.; Rachubinski, R.A.; Watkins, S.C.; Billiar, T.R. PEX7 and EBP50 target iNOS to the peroxisome in hepatocytes. Nitric Oxide 2013, 31, 9–19.
- Prolo, C.; Alvarez, M.N.; Radi, R. Peroxynitrite, a potent macrophage-derived oxidizing cytotoxin to combat invading pathogens. Biofactors 2014, 40, 215–225.
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837.
- Lacza, Z.; Puskar, M.; Figueroa, J.P.; Zhang, J.; Rajapakse, N.; Busija, D.W. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic Biol. Med. 2001, 31, 1609–1615.
- Ghafourifar, P.; Cadenas, E. Mitochondrial nitric oxide synthase. Trends Pharmacol. Sci. 2005, 26, 190–195.
- Nisoli, E.; Tonello, C.; Cardile, A.; Cozzi, V.; Bracale, R.; Tedesco, L.; Falcone, S.; Valerio, A.; Cantoni, O.; Clementi, E.; et al. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 2005, 310, 314–317.
- Zhu, L.; Wang, Q.; Zhang, L.; Fang, Z.; Zhao, F.; Lv, Z.; Gu, Z.; Zhang, J.; Wang, J.; Zen, K.; et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010, 20, 676–687.
- Victor, V.M.; Nuñez, C.; D’Ocón, P.; Taylor, C.T.; Esplugues, J.V.; Moncada, S. Regulation of oxygen distribution in tissues by endothelial nitric oxide. Circ. Res. 2009, 104, 1178–1183.
- Moens, A.L.; Kass, D.A. Tetrahydrobiopterin and cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2439–2444.
- Garcin, E.D.; Bruns, C.M.; Lloyd, S.J.; Hosfield, D.J.; Tiso, M.; Gachhui, R.; Stuehr, D.J.; Tainer, J.A.; Getzoff, E.D. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J. Biol. Chem. 2004, 279, 37918–37927.
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531.
- Rubio-Guerra, A.F.; Vargas-Robles, H.; Ramos-Brizuela, L.M.; Escalante-Acosta, B.A. Is tetrahydrobiopterin a therapeutic option in diabetic hypertensive patients? Integr. Blood Press. Control 2010, 3, 125–132.
- Landmesser, U.; Dikalov, S.; Price, S.R.; McCann, L.; Fukai, T.; Holland, S.M.; Mitch, W.E.; Harrison, D.G. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Investig. 2003, 111, 1201–1209.
- Hoang, H.H.; Padgham, S.V.; Meininger, C.J. L-arginine, tetrahydrobiopterin, nitric oxide and diabetes. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 76–82.
- Gámez-Méndez, A.M.; Vargas-Robles, H.; Arellano-Mendoza, M.; Cruz-Laguna, E.; Rios, A.; Escalante, B. Early stage of obesity potentiates nitric oxide reduction during the development of renal failure. J. Nephrol. 2014, 27, 281–287.
- Rabender, C.S.; Alam, A.; Sundaresan, G.; Cardnell, R.J.; Yakovlev, V.A.; Mukhopadhyay, N.D.; Graves, P.; Zweit, J.; Mikkelsen, R.B. The Role of Nitric Oxide Synthase Uncoupling in Tumor Progression. Mol. Cancer Res. 2015, 13, 1034–1043.
- Veron, D.; Aggarwal, P.K.; Velazquez, H.; Kashgarian, M.; Moeckel, G.; Tufro, A. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J. Am. Soc. Nephrol. 2014, 25, 1814–1824.
- Seo, B.R.; Bhardwaj, P.; Choi, S.; Gonzalez, J.; Andresen Eguiluz, R.C.; Wang, K.; Mohanan, S.; Morris, P.G.; Du, B.; Zhou, X.K.; et al. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci. Transl. Med. 2015, 7, 301ra130.
- Edgar, K.S.; Matesanz, N.; Gardiner, T.A.; Katusic, Z.S.; McDonald, D.M. Hyperoxia depletes (6R)-5,6,7,8-tetrahydrobiopterin levels in the neonatal retina: Implications for nitric oxide synthase function in retinopathy. Am. J. Pathol. 2015, 185, 1769–1782.
- Kuzkaya, N.; Weissmann, N.; Harrison, D.G.; Dikalov, S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: Implications for uncoupling endothelial nitric-oxide synthase. J. Biol. Chem. 2003, 278, 22546–22554.
- Werner-Felmayer, G.; Golderer, G.; Werner, E.R. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Curr. Drug Metab. 2002, 3, 159–173.
- Kim, H.L.; Park, Y.S. Maintenance of cellular tetrahydrobiopterin homeostasis. BMB Rep. 2010, 43, 584–592.
- Latremoliere, A.; Costigan, M. GCH1, BH4 and pain. Curr. Pharm. Biotechnol. 2011, 12, 1728–1741.
- Shintaku, H. Disorders of tetrahydrobiopterin metabolism and their treatment. Curr. Drug Metab. 2002, 3, 123–131.
- Martínez-Ruiz, A.; Araújo, I.M.; Izquierdo-Álvarez, A.; Hernansanz-Agustín, P.; Lamas, S.; Serrador, J. Specificity in S-nitrosylation: A short-range mechanism for NO signaling? Antioxid. Redox Signal. 2013, 19, 1220–1235.
- Figueroa, X.F.; Lillo, M.A.; Gaete, P.S.; Riquelme, M.A.; Sáez, J.C. Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin-based channel. Neuropharmacology 2013, 75, 471–478.
- Nakamura, T.; Lipton, S.A. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid. Redox Signal. 2013, 18, 239–249.
- Chang, A.H.; Sancheti, H.; Garcia, J.; Kaplowitz, N.; Cadenas, E.; Han, D. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem. Res. Toxicol. 2014, 27, 794–804.
- Piantadosi, C.A. Regulation of Mitochondrial Processes by Protein S-Nitrosylation. Biochim. Biophys. Acta 2012, 1820, 712–721.
- Clementi, E.; Brown, G.C.; Feelisch, M.; Moncada, S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA 1998, 95, 7631–7636.
- Penna, C.; Sorge, M.; Femminò, S.; Pagliaro, P.; Brancaccio, M. Redox Aspects of Chaperones in Cardiac Function. Front. Physiol. 2018, 9, 216.
- Rossi-George, A.; Gow, A. Nitric Oxide Biochemistry: Pathophysiology of Nitric Oxide-Mediated Protein Modifications. In Oxidative Neural Injury, Contemporary Clinical Neuroscience; Veasey, S.C., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 29–44.
- Oae, S.; Shinhama, K. Organic thionitrites and related substances. Org. Prep. Proced. Int. 1983, 16, 165–198.
- Stamler, J.S.; Jaraki, O.; Osborne, J.; Simon, D.I.; Keaney, J.; Vita, J.; Singel, D.; Valeri, C.R.; Loscalzo, J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc. Natl. Acad. Sci. USA 1992, 88, 7674–7677.
- Anand, P.; Stamler, J. Enzymatic mechanisms regulating protein S-nitrosylation: Implications in health and disease. J. Mol. Med. (Berl.) 2012, 90, 233–244.
- Keilin, D.; Hartree, E.F. Reaction of nitric oxide with haemoglobin and methaemoglobin. Nature 1937, 139, 548.
- Murad, F. Regulation of cytosolic guanylyl cyclase by nitric oxide: The NO- cyclic GMP signal transduction system. Adv. Pharmacol. 1994, 26, 19–33.
- Sarti, P.; Forte, E.; Mastronicola, D.; Giuffrè, A.; Arese, M. Cytochrome c oxidase and nitric oxide in action: Molecular mechanisms and pathophysiological implications. Biochim. Biophys. Acta 2012, 1817, 610–619.
- Fago, A.; Crumbliss, A.L.; Hendrich, M.P.; Pearce, L.L.; Peterson, J.; Henkens, R.; Bonaventura, C. Oxygen binding to partially nitrosylated hemoglobin. Biochim. Biophys. Acta 2013, 1834, 1894–1900.
- Jung, T.; Höhn, A.; Grune, T. The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biol. 2014, 2, 99–104.
- Freeman, B.A.; Baker, P.R.; Schopfer, F.J.; Woodcock, S.R.; Napolitano, A.; d’Ischia, M. Nitro-fatty acid formation and signaling. J. Biol. Chem. 2008, 283, 15515–15519.
- Lancaster, J.R.J. Nitric oxide: A brief overview of chemical and physical properties relevant to therapeutic applications. Future Sci. OA 2015, 1, FSO59.
- Ravi, K.; Brennan, L.A.; Levic, S.; Ross, P.A.; Black, S.M. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc. Natl. Acad. Sci. USA 2004, 101, 2619–2624.
- Thapliyal, A.; Verma, R.; Kumar, N. Small G Proteins Dexras1 and RHES and Their Role in Pathophysiological Processes. Int. J. Cell Biol. 2014, 2014, 308535.
- Trepel, J.; Mollapour, M.; Giaccone, G.; Neckers, L. Targeting the dynamic HSP90 complex in cancer. Nat. Rev. Cancer 2010, 10, 537–549.
- Hsu, K.; Champaiboon, C.; Guenther, B.D.; Sorenson, B.S.; Khammanivong, A.; Ross, K.F.; Geczy, C.L.; Herzberg, M.C. Anti-infective protective properties of s100 calgranulins. Anti Inflamm. Anti Allergy Agents Med. Chem. 2009, 8, 290–305.
- Jia, J.; Arif, A.; Terenzi, F.; Willard, B.; Plow, E.F.; Hazen, S.L.; Fox, P.L. Target-selective protein S-nitrosylation by sequence motif recognition. Cell 2014, 159, 623–634.
- Lim, S.Y.; Raftery, M.; Cai, H.; Hsu, K.; Yan, W.X.; Hseih, H.L.; Watts, R.N.; Richardson, D.; Thomas, S.; Perry, M.; et al. S-nitrosylated S100A8: Novel anti-inflammatory properties. J. Immunol. 2008, 181, 5627–5636.
- Mendez, M.G.; Kojima, S.; Goldman, R.D. JVimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB 2010, 26, 1838–1851.
- Fehon, R.G.; McClatchey, A.; Bretscher, A. Organizing the cell cortex: The role of ERM proteins. Nat. Rev. Mol. Cell Biol. 2010, 11, 276–287.
- Pérez-Mato, I.; Castro, C.; Ruiz, F.A.; Corrales, F.J.; Mato, J.M. Methionine adenosyltransferase S-nitrosylation is regulated by the basic and acidic amino acids surrounding the target thiol. J. Biol. Chem. 1999, 274, 17075–17079.
- Doulias, P.T.; Greene, J.L.; Greco, T.M.; Tenopoulou, M.; Seeholzer, S.H.; Dunbrack, R.L.; Ischiropoulos, H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc. Natl. Acad. Sci. USA 2010, 107, 16958–16963.
- Möller, M.N.; Li, Q.; Vitturi, D.A.; Robinson, J.M.; Lancaster, J.R.J.; Denicola, A. Membrane “lens” effect: Focusing the formation of reactive nitrogen oxides from the *NO/O2 reaction. Chem. Res. Toxicol. 2007, 20, 709–714.
- Broniowska, K.A.; Hogg, N. The chemical biology of S-nitrosothiols. Antioxid. Redox Signal. 2012, 17, 969–980.
- Bartberger, M.D.; Liu, W.; Ford, E.; Miranda, K.M.; Switzer, C.; Fukuto, J.M.; Farmer, P.J.; Wink, D.A.; Houk, K.N. The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proc. Natl. Acad. Sci. USA 2002, 99, 10958–10963.
- Cheng, S.; Shi, T.; Wang, X.L.; Liang, J.; Wu, H.; Xie, L.; Li, Y.; Zhao, Y.L. Features of S-nitrosylation based on statistical analysis and molecular dynamics simulation: Cysteine acidity, surrounding basicity, steric hindrance and local flexibility. Mol. Biosyst. 2014, 10, 2597–2606.
- Beltrán, B.; Orsi, A.; Clementi, E.; Moncada, S. Oxidative stress and S-nitrosylation of proteins in cells. Br. J. Pharmacol. 2000, 129, 953–960.
- Butzer, U.; Weidenbach, H.; Gansauge, S.; Gansauge, F.; Beger, H.G.; Nussler, A.K. Increased oxidative stress in the RAW 264.7 macrophage cell line is partially mediated via the S-nitrosothiol-induced inhibition of glutathione reductase. FEBS Lett. 1999, 445, 274–278.
- Whiteman, M.; Chua, Y.L.; Zhang, D.; Duan, W.; Liou, Y.C.; Armstrong, J.S. Nitric oxide protects against mitochondrial permeabilization induced by glutathione depletion: Role of S-nitrosylation? Biochem. Biophys. Res. Commun. 2006, 339, 255–262.
- Stomberski, C.T.; Hess, D.T.; Stamler, J.S. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxid. Redox Signal. 2019, 30, 1331–1351.
- Kovacs, I.; Holzmeister, C.; Wirtz, M.; Geerlof, A.; Fröhlich, T.; Römling, G.; Kuruthukulangarakoola, G.T.; Linster, E.; Hell, R.; Arnold, G.J.; et al. ROS-Mediated Inhibition of S-nitrosoglutathione Reductase Contributes to the Activation of Anti-oxidative Mechanisms. Front. Plant Sci. 2016, 7, 1669.
- Zhang, Y.; Wu, K.; Su, W.; Zhang, D.F.; Wang, P.; Qiao, X.; Yao, Q.; Yuan, Z.; Yao, Y.G.; Liu, G.; et al. Increased GSNOR Expression during Aging Impairs Cognitive Function and Decreases S-Nitrosation of CaMKIIα. J. Neurosci. 2017, 37, 9741–9758.
- Caplin, B.; Leiper, J. Endogenous nitric oxide synthase inhibitors in the biology of disease: Markers, mediators, and regulators? Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1343–1353.
- Fan, J.S.; Zhang, Q.; Li, M.; Tochio, H.; Yamazaki, T.; Shimizu, M.; Zhang, M. Protein inhibitor of neuronal nitric-oxide synthase, PIN, binds to a 17-amino acid residue fragment of the enzyme. J. Biol. Chem. 1998, 273, 33472–33481.
- Broniowska, K.A.; Keszler, A.; Basu, S.; Kim-Shapiro, D.B.; Hogg, N. Cytochrome c-mediated formation of S-nitrosothiol in cells. Biochem. J. 2012, 442, 191–197.
- Dahm, C.C.; Moore, K.; Murphy, M.P. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: Implications for the interaction of nitric oxide with mitochondria. J. Biol. Chem. 2006, 281, 10056–10065.
- Giri, S.; Rattan, R.; Deshpande, M.; Maguire, J.L.; Johnson, Z.; Graham, R.P.; Shridhar, V. Preclinical therapeutic potential of a nitrosylating agent in the treatment of ovarian cancer. PLoS ONE 2014, 9, e97897.
- Furuhashi, S.; Sugita, H.; Takamori, H.; Horino, K.; Nakahara, O.; Okabe, H.; Miyake, K.; Tanaka, H.; Beppu, T.; Baba, H. NO donor and MEK inhibitor synergistically inhibit proliferation and invasion of cancer cells. Int. J. Oncol. 2012, 40, 807–815.
- Olson, N.; Kasahara, D.; Hristova, M.; Bernstein, R.; Janssen-Heininger, Y.; van der Vliet, A. Modulation of NF-κB and hypoxia-inducible factor--1 by S-nitrosoglutathione does not alter allergic airway inflammation in mice. Am. J. Respir. Cell Mol. Biol. 2011, 44, 813–823.
- Kaliyaperumal, K.; Sharma, A.K.; McDonald, D.G.; Dhindsa, J.S.; Yount, C.; Singh, A.K.; Won, J.S.; Singh, I. S-Nitrosoglutathione-mediated STAT3 regulation in efficacy of radiotherapy and cisplatin therapy in head and neck squamous cell carcinoma. Redox Biol. 2015, 6, 41–50.
- Marozkina, N.V.; Gaston, B. S-Nitrosylation signaling regulates cellular protein interactions. Biochim. Biophys. Acta 2012, 1820, 722–729.
- Karin, M. NF-κB as a Critical Link Between Inflammation and Cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141.
- Tristan, C.; Shahani, N.; Sedlak, T.W.; Sawa, A. The diverse functions of GAPDH: Views from different subcellular compartments. Cell. Signal. 2011, 23, 317–323.
- Hara, M.R.; Agrawal, N.; Kim, S.F.; Cascio, M.B.; Fujimuro, M.; Ozeki, Y.; Takahashi, M.; Cheah, J.H.; Tankou, S.K.; Hester, L.D.; et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 2005, 7, 665–674.
- Kornberg, M.D.; Sen, N.; Hara, M.R.; Juluri, K.R.; Nguyen, J.V.; Snowman, A.M.; Law, L.; Hester, L.D.; Snyder, S.H. GAPDH mediates nitrosylation of nuclear proteins. Nat. Cell Biol. 2010, 12, 1094–1100.
- Reed, J.C.; Ely, K.R. Degrading liaisons: Siah structure revealed. Nat. Struct. Biol. 2002, 9, 8–10.
- Zhai, D.; Chin, K.; Wang, M.; Liu, F. Disruption of the nuclear p53-GAPDH complex protects against ischemia-induced neuronal damage. Mol. Brain 2014, 7, 20.
- Thangima Zannat, M.; Bhattacharjee, R.B.; Bag, J. In the absence of cellular poly (A) binding protein, the glycolytic enzyme GAPDH translocated to the cell nucleus and activated the GAPDH mediated apoptotic pathway by enhancing acetylation and serine 46 phosphorylation of p53. Biochem. Biophys. Res. Commun. 2011, 409, 171–176.
- Kohr, M.J.; Murphy, E.; Steenbergen, C. Glyceraldehyde-3-phosphate dehydrogenase acts as a mitochondrial trans-S-nitrosylase in the heart. PLoS ONE 2014, 9, e111448.
- Nakajima, H.; Itakura, M.; Kubo, T.; Kaneshige, A.; Harada, N.; Izawa, T.; Azuma, Y.T.; Kuwamura, M.; Yamaji, R.; Takeuchi, T. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Aggregation Causes Mitochondrial Dysfunction during Oxidative Stress-induced Cell Death. J. Biol. Chem. 2017, 292, 4727–4742.
- Wen, Y.Y.; Yang, Z.Q.; Song, M.; Li, B.L.; Yao, X.H.; Chen, X.L.; Zhao, J.; Lu, Y.Y.; Zhu, J.J.; Wang, E.H. The expression of SIAH1 is downregulated and associated with Bim and apoptosis in human breast cancer tissues and cells. Mol. Carcinog. 2010, 49, 440–449.
- Zhang, J.Y.; Zhang, F.; Hong, C.Q.; Giuliano, A.E.; Cui, X.J.; Zhou, G.J.; Zhang, G.J.; Cui, Y.K. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol. Med. 2015, 12, 10–22.
- Fiucci, G.; Beaucourt, S.; Duflaut, D.; Lespagnol, A.; Stumptner-Cuvelette, P.; Géant, A.; Buchwalter, G.; Tuynder, M.; Susini, L.; Lassalle, J.M.; et al. Siah-1b is a direct transcriptional target of p53: Identification of the functional p53 responsive element in the siah-1b promote. Proc. Natl. Acad. Sci. USA 2004, 101, 3510–3515.
- Tardif, M.R.; Chapeton-Montes, J.A.; Posvandzic, A.; Pagé, N.; Gilbert, C.; Tessier, P.A. Secretion of S100A8, S100A9, and S100A12 by Neutrophils Involves Reactive Oxygen Species and Potassium Efflux. J. Immunol. Res. 2015, 2015, 296149.
- Kerkhoff, C.; Nacken, W.; Benedyk, M.; Dagher, M.C.; Sopalla, C.; Doussiere, J. The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. FASAB J. 2005, 19, 467–469.
- Biri-Kovács, B.; Kiss, B.; Vadászi, H.; Gógl, G.; Pálfy, G.; Török, G.; Homolya, L.; Bodor, A.; Nyitray, L. Ezrin interacts with S100A4 via both its N- and C-terminal domains. PLoS ONE 2017, 12, e0177489.
- Baritaki, S.; Huerta-Yepez, S.; Sahakyan, A.; Karagiannides, I.; Bakirtzi, K.; Jazirehi, A.; Bonavida, B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: Inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle 2010, 9, 4931–4940.
- Mannick, J.B.; Schonhoff, C.; Papeta, N.; Ghafourifar, P.; Szibor, M.; Fang, K.; Gaston, B. S-Nitrosylation of mitochondrial caspases. J. Cell Biol. 2001, 154, 1111–1116.
- Santhanam, L.; Tuday, E.C.; Webb, A.K.; Dowzicky, P.; Kim, J.H.; Oh, Y.J.; Sikka, G.; Kuo, M.; Halushka, M.K.; Macgregor, A.M.; et al. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ. Res. 2010, 107, 117–125.
- Telci, D.; Collighan, R.J.; Basaga, H.; Griffin, M. Increased TG2 expression can result in induction of transforming growth factor beta1, causing increased synthesis and deposition of matrix proteins, which can be regulated by nitric oxide. J. Biol. Chem. 2009, 284, 29547–29558.
- Tang, C.H.; Wei, W.; Liu, L. Regulation of DNA repair by S-nitrosylation. Biochim. Biophys. Acta 2012, 1820, 730–735.
- Benhar, M. Emerging Roles of Protein S-Nitrosylation in Macrophages and Cancer Cells. Curr. Med. Chem. 2016, 23, 2602–2617.
- Nott, A.; Nitarska, J.; Veenvliet, J.V.; Schacke, S.; Derijck, A.A.H.A.; Sirko, P.; Muchardt, C.; Pasterkamp, R.J.; Smidt, M.P.; Riccio, A. S-nitrosylation of HDAC2 regulates the expression of the chromatin-remodeling factor Brm during radial neuron migration. Proc. Natl. Acad. Sci. USA 2013, 110, 3113–3118.
- Nott, A.; Watson, P.M.; Robinson, J.D.; Crepaldi, L.; Riccio, A. S-nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature 2008, 455, 411.
- Gu, Z.; Kaul, M.; Yan, B.; Kridel, S.J.; Cui, J.; Strongin, A.; Smith, J.W.; Liddington, R.C.; Lipton, S.A. S-nitrosylation of matrix metalloproteinases: Signaling pathway to neuronal cell death. Science 2002, 297, 1186–1190.
- O’Sullivan, S.; Medina, C.; Ledwidge, M.; Radomski, M.W.; Gilmer, J.F. Nitric oxide-matrix metaloproteinase-9 interactions: Biological and pharmacological significance--NO and MMP-9 interactions. Biochim. Biophys. Acta 2014, 1843, 603–617.
- Bignon, E.; Allega, M.F.; Lucchetta, M.; Tiberti, M.; Papaleo, E. Computational Structural Biology of S-nitrosylation of Cancer Targets. Front. Oncol. 2018, 8, 272.
- Thibeault, S.; Rautureau, Y.; Oubaha, M.; Faubert, D.; Wilkes, B.C.; Delisle, C.; Gratton, J.P. S-Nitrosylation of β-Catenin by eNOS-Derived NO Promotes VEGF-Induced Endothelial Cell Permeability. Mol. Cell 2010, 39, 468–476.
- Nakamura, T.; Prikhodko, O.A.; Pirie, E.; Nagar, S.; Akhtar, M.W.; Oh, C.K.; McKercher, S.R.; Ambasudhan, R.; Okamoto, S.I.; Lipton, S.A. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol. Dis. 2015, 84, 99–108.
- Casero, R.A.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695.
- Zhu, L.; Zhang, C.; Liu, Q. PTEN S-nitrosylation by NOS1 inhibits autophagy in NPC cells. Cell Death Dis. 2019, 10, 306.
- Marshall, H.E.; Foster, M.W. S-nitrosylation of Ras in breast cancer. Breast Cancer Res. 2012, 14, 113.
- Robb, E.L.; Page, M.M.; Stuart, J.A. Mitochondria, cellular stress resistance, somatic cell depletion and lifespan. Curr. Aging Sci. 2012, 2, 12–27.
- Nakamura, T.; Lipton, S. Cell death: Protein misfolding and neurodegenerative diseases. Apoptosis 2009, 14, 455–468.
- Ozawa, K.; Komatsubara, A.T.; Nishimura, Y.; Sawada, T.; Kawafune, H.; Tsumoto, H.; Tsuji, Y.; Zhao, J.; Kyotani, Y.; Tanaka, T.; et al. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci. Rep. 2013, 3, 2202.
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2013, 2, 129–139.
- Doulias, P.T.; Tenopoulou, M.; Greene, J.L.; Raju, K.; Ischiropoulos, H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci. Signal. 2013, 6, rs1.
- Nguyen, T.T.; Stevens, M.V.; Kohr, M.; Steenbergen, C.; Sack, M.N.; Murphy, E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J. Biol. Chem. 2011, 286, 40184–40192.
- Azad, N.; Vallyathan, V.; Wang, L.; Tantishaiyakul, V.; Stehlik, C.; Leonard, S.S.; Rojanasakul, Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. J. Biol. Chem. 2006, 281, 34124–34134.
- Brown, G.C.; Borutaite, V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim. Biophys. Acta 2004, 1658, 44–49.
- Hroudová, J.; Fišar, Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regen. Res. 2013, 8, 363–375.
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13.
- Zhang, J.; Jin, B.; Li, L.; Block, E.R.; Patel, J.M. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am. J. Physiol. Cell Physiol. 2005, 288, C840–C849.
- Le Cras, T.D.; McMurtry, I.F. Nitric oxide production in the hypoxic lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L575–L582.
- Hagen, T.; Taylor, C.T.; Lam, F.; Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: Effect on HIF1alpha. Science 2003, 302, 1975–1978.
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59.
- Sack, M.N. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc. Res. 2006, 72, 210–219.
- Basu, S.; Keszler, A.; Azarova, N.A.; Nwanze, N.; Perlegas, A.; Shiva, S.; Broniowska, K.A.; Hogg, N.; Kim-Shapiro, D.B. A novel role for cytochrome c: Efficient catalysis of S-nitrosothiol formation. Free Radic Biol. Med. 2010, 48, 255–263.
- Schonhoff, C.M.; Gaston, B.; Mannick, J.B. Nitrosylation of cytochrome c during apoptosis. J. Biol. Chem. 2003, 278, 18265–18270.
- Liemburg-Apers, D.C.; Willems, P.H.; Koopman, W.J.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226.
- Speijer, D.; Manjeri, G.R.; Szklarczyk, R. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130446.
- Quijano, C.; Trujillo, M.; Castro, L.; Trostchansky, A. Interplay between oxidant species and energy metabolism. Redox Biol. 2016, 8, 28–42.
- Quinlan, C.L.; Perevoshchikova, I.V.; Hey-Mogensen, M.; Orr, A.L.; Brand, M.D. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013, 1, 304–312.
- Chouchani, E.T.; Hurd, T.R.; Nadtochiy, S.M.; Brookes, P.S.; Fearnley, I.M.; Lilley, K.S.; Smith, R.A.; Murphy, M.P. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): Implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem. J. 2010, 430, 49–59.
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401.
- Nguyen, T.T.M.; Stevens, M.; Kohr, M.; Steenbergen, C.; Sack, M.; Murphy, E. S-nitrosylation of cyclophilin D alters mitochondrial permeability transition pore. FASEB J. 2011, 25, 1031–1033.
- Ohtani, H.; Katoh, H.; Tanaka, T.; Saotome, M.; Urushida, T.; Satoh, H.; Hayashi, H. Effects of nitric oxide on mitochondrial permeability transition pore and thiol-mediated responses in cardiac myocytes. Nitric Oxide 2012, 26, 95–101.
- Shoshan-Barmatz, V.; De Pinto, V.; Zweckstetter, M.; Raviv, Z.; Keinan, N.; Arbel, N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol. Asp. Med. 2010, 31, 227–285.
- Cheng, Q.; Sedlic, F.; Pravdic, D.; Bosnjak, Z.J.; Kwok, W.M. Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channe. FEBS Lett. 2011, 585, 328–334.
- Crow, M.T.; Mani, K.; Nam, Y.J.; Kitsis, R.N. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ. Res. 2004, 95, 957–970.
- Gobeil, F.J.; Zhu, T.; Brault, S.; Geha, A.; Vazquez-Tello, A.; Fortier, A.; Barbaz, D.; Checchin, D.; Hou, X.; Nader, M.; et al. Nitric oxide signaling via nuclearized endothelial nitric-oxide synthase modulates expression of the immediate early genes iNOS and mPGES-1. J. Biol. Chem. 2006, 281, 16058–16067.
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Nuclear recruitment of neuronal nitric-oxide synthase by α-syntrophin is crucial for the induction of mitochondrial biogenesis. J. Biol. Chem. 2014, 289, 365–378.
- Jones, R.J.; Jourd’heuil, D.; Salerno, J.C.; Smith, S.M.; Singer, H.A. iNOS regulation by calcium/calmodulin-dependent protein kinase II in vascular smooth muscle. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H2634–H2642.
- Villanueva, C.; Giulivi, C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol. Med. 2010, 49, 307–316.
- Raghunath, A.; Sundarraj, K.; Nagarajan, R.; Arfuso, F.; Bian, J.; Kumar, A.P.; Sethi, G.; Perumal, E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018, 17, 297–314.
- Teixeira, T.M.; da Costa, D.C.; Resende, A.C.; Soulage, C.O.; Bezerra, F.F.; Daleprane, J.B. Activation of Nrf2-Antioxidant Signaling by 1,25-Dihydroxycholecalciferol Prevents Leptin-Induced Oxidative Stress and Inflammation in Human Endothelial Cells. J. Nutr. 2017, 47, 506–513.
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745.
- Um, H.C.; Jang, J.H.; Kim, D.H.; Lee, C.; Surh, Y.J. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide 2011, 25, 161–168.
- Aquilano, K.; Baldelli, S.; Pagliei, B.; Cannata, S.M.; Rotilio, G.; Ciriolo, M.R. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid. Redox Signal. 2013, 18, 386–399.
- Baldelli, S.; Ciriolo, M.R. Altered S-nitrosylation of p53 is responsible for impaired antioxidant response in skeletal muscle during aging. Aging (Albany NY) 2016, 8, 3450–3467.
- Cherry, A.D.; Suliman, H.B.; Bartz, R.R.; Piantadosi, C.A. Peroxisome proliferator-activated receptor γ co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. J. Biol. Chem. 2014, 289, 41–52.
- Schonhoff, C.M.; Daou, M.C.; Jones, S.N.; Schiffer, C.A.; Ross, A.H. Nitric oxide-mediated inhibition of Hdm2-p53 binding. Biochemistry 2002, 41, 13570–13574.
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626.
- Gloire, G.; Legrand-Poels, S.; Piette, J. NF-kappaB activation by reactive oxygen species: Fifteen years later. Biochem. Pharmacol. 2006, 72, 1493–1550.
- Reynaert, N.L.; Ckless, K.; Korn, S.H.; Vos, N.; Guala, A.S.; Wouters, E.F.; van der Vliet, A.; Janssen-Heininger, Y.M. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc. Natl. Acad. Sci. USA 2004, 101, 8945–8950.
- Mauro, C.; Leow, S.C.; Anso, E.; Rocha, S.; Thotakura, A.K.; Tornatore, L.; Moretti, M.; De Smaele, E.; Beg, A.A.; Tergaonkar, V.; et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011, 13, 1272–1279.
- Semenza, G.L. Hydroxylation of HIF-1: Oxygen sensing at the molecular level. Physiology 2004, 19, 176–182.
- Brune, B.; Zhou, J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1α (HIF-1α). Curr. Med. Chem. 2003, 10, 845–855.
- Terry, L.J.; Shows, E.B.; Wente, S.R. Crossing the nuclear envelope: Hierarchical regulation of nucleocytoplasmic transport. Science 2007, 318, 1412–1416.
- Fornerod, M.; Ohno, M.; Yoshida, M.; Mattaj, I.W. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 1997, 90, 1051–1060.
- Fu, S.C.; Huang, H.C.; Horton, P.; Juan, H.F. ValidNESs: A database of validated leucine-rich nuclear export signals. Nucleic Acids Res. 2013, 41, D338–D343.
- Mutka, S.C.; Yang, W.Q.; Dong, S.D.; Ward, S.L.; Craig, D.A.; Timmermans, P.B.; Murli, S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009, 69, 510–517.
- Wang, P.; Liu, G.H.; Wu, K.; Qu, J.; Huang, B.; Zhang, X.; Zhou, X.; Gerace, L.; Chen, C. Repression of classical nuclear export by S-nitrosylation of CRM1. J. Cell Sci. 2009, 122 Pt 20, 3772–3779.
- Freedman, D.A.; Levine, A.J. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol. Cell. Biol. 1998, 18, 7288–7293.
- Forrester, M.T.; Thompson, J.W.; Foster, M.W.; Nogueira, L.; Moseley, M.A.; Stamler, J.S. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat. Biotechnol. 2009, 27, 557–559.
- Azam, S.; Jouvet, N.; Jilani, A.; Vongsamphanh, R.; Yang, X.; Yang, S.; Ramotar, D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J. Biol. Chem. 2008, 283, 30632–30641.
- Dobbs, T.A.; Tainer, J.A.; Lees-Miller, S.P. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst.) 2010, 9, 1307–1314.
- Xu, W.; Liu, L.; Smith, G.C.; Charles, l.G. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging anti-tumour agents. Nat. Cell Biol. 2000, 2, 339–345.
- Tell, G.; Quadrifoglio, F.; Tiribelli, C.; Kelley, M.R. The many functions of APE1/Ref-1: Not only a DNA repair enzyme. Antioxid. Redox Signal. 2009, 11, 601–620.
- Choi, S.; Joo, H.K.; Jeon, B.H. Dynamic Regulation of APE1/Ref-1 as a Therapeutic Target Protein. Chonnam Med. J. 2016, 52, 75–80.
- Qu, J.; Liu, G.H.; Huang, B.; Chen, C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007, 35, 2522–2532.
- Ottaviano, F.G.; Handy, D.E.; Loscalzo, J. Redox regulation in the extracellular environment. Circ. J. 2008, 72, 1–16.
- Stamler, J.S.; Lamas, S.; Fang, F.C. Nitrosylation. the prototypic redox-based signaling mechanism. Cell 2001, 106, 675–683.
- Koh, Y.H.; Suzuki, K.; Che, W.; Park, Y.S.; Miyamoto, Y.; Higashiyama, S.; Taniguchi, N. Inactivation of glutathione peroxidase by NO leads to the accumulation of H2O2 and the induction of HB-EGF via c-Jun NH2-terminal kinase in rat aortic smooth muscle cells. FASEB J. 2001, 15, 1472–1474.
- Ortega-Galisteo, A.P.; Rodríguez-Serrano, M.; Pazmiño, D.M.; Gupta, D.K.; Sandalio, L.M.; Romero-Puertas, M.C. S-Nitrosylated proteins in pea (Pisum sativum L.) leaf peroxisomes: Changes under abiotic stress. J. Exp. Bot. 2012, 63, 2089–2103.
- Furuta, S. Basal S-Nitrosylation Is the Guardian of Tissue Homeostasis. Trends Cancer 2017, 3, 744–748.
- Subczynski, W.K.; Lomnicka, M.; Hyde, J.S. Permeability of nitric oxide through lipid bilayer membranes. Free Radic Res. 1996, 24, 343–349.
- Herrera, M.; Hong, N.J.; Garvin, J.L. Aquaporin-1 transports NO across cell membranes. Hypertension 2006, 48, 157–164.
- Denicola, A.; Souza, J.M.; Radi, R.; Lissi, E. Nitric oxide diffusion in membranes determined by fluorescence quenching. Arch. Biochem. Biophys. 1996, 328, 208–212.
- Malysheva, E.V.; Kruglov, S.V.; Khomenko, I.P.; Bakhtina, L.Y.; Pshennikova, M.G.; Manukhina, E.B.; Malyshev, I.Y. Role of extracellular and intracellular nitric oxide in the regulation of macrophage responses. Bull. Exp. Biol. Med. 2006, 141, 404–406.
- Matsumoto, A.; Gow, A.J. Membrane transfer of S-nitrosothiols. Nitric Oxide 2011, 25, 102–107.
- Iwakiri, Y.; Satoh, A.; Chatterjee, S.; Toomre, D.K.; Chalouni, C.M.; Fulton, D.; Groszmann, R.; Shah, V.H.; Sessa, W.C. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc. Natl. Acad. Sci. USA 2006, 103, 19777–19782.
- Sangwung, P.; Greco, T.M.; Wang, Y.; Ischiropoulos, H.; Sessa, W.C.; Iwakiri, Y. Proteomic identification of S-nitrosylated Golgi proteins: New insights into endothelial cell regulation by eNOS-derived NO. PLoS ONE 2012, 7, e31564.
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604.
- Hess, D.T.; Matsumoto, A.; Nudelman, R.; Stamler, J.S. S-nitrosylation: Spectrum and specificity. Nat. Cell Biol. 2001, 3, E46–E48.
- Savage, D.F.; Stroud, R.M. Structural basis of aquaporin inhibition by mercury. J. Mol. Biol. 2007, 368, 607–617.
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313.
- Matono, R.; Miyano, K.; Kiyohara, T.; Sumimoto, H. Arachidonic acid induces direct interaction of the p67(phox)-Rac complex with the phagocyte oxidase Nox2, leading to superoxide production. J. Biol. Chem. 2014, 289, 24874–24884.
- Lou, Z.; Wang, A.P.; Duan, X.M.; Hu, G.H.; Song, G.L.; Zuo, M.L.; Yang, Z.B. Upregulation of NOX2 and NOX4 Mediated by TGF-β Signaling Pathway Exacerbates Cerebral Ischemia/Reperfusion Oxidative Stress Injury. Cell Physiol. Biochem. 2018, 46, 2103–2113.
- Selemidis, S.; Dusting, G.; Peshavariya, H.; Kemp-Harper, B.K.; Drummond, G.R. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc. Res. 2007, 75, 349–358.
- Harrison, C.B.; Drummond, G.R.; Sobey, C.G.; Selemidis, S. Evidence that nitric oxide inhibits vascular inflammation and superoxide production via a p47phox-dependent mechanism in mic. Clin. Exp. Pharmacol. Physiol. 2010, 37, 429–434.
- Schmidt, H.M.; Kelley, E.E.; Straub, A.C. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019, 21, 101072.
- White, C.R. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc. Natl. Acad. Sci. USA 1996, 93, 8745–8749.
- Khan, S.A.; Lee, K.; Minhas, K.M.; Gonzalez, D.R.; Raju, S.V.; Tejani, A.D.; Li, D.; Berkowitz, D.E.; Hare, J.M. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc. Natl. Acad. Sci. USA 2004, 101, 15944–15948.
- Saraiva, R.M.; Minhas, K.M.; Zheng, M.; Pitz, E.; Treuer, A.; Gonzalez, D.; Schuleri, K.H.; Vandegaer, K.M.; Barouch, L.A.; Hare, J.M. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric Oxide 2007, 16, 331–338.
- Petersen, S.V.; Oury, T.D.; Ostergaard, L.; Valnickova, Z.; Wegrzyn, J.; Thøgersen, I.B.; Jacobsen, C.; Bowler, R.P.; Fattman, C.L.; Crapo, J.D.; et al. Extracellular superoxide dismutase (EC-SOD) binds to type i collagen and protects against oxidative fragmentation. J. Biol. Chem. 2004, 279, 13705–13710.
- Jung, O.; Marklund, S.L.; Geiger, H.; Pedrazzini, T.; Busse, R.; Brandes, R.P. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: In vivo and ex vivo evidence from ecSOD-deficient mice. Circ. Res. 2003, 93, 622–629.
- Holzmeister, C.; Gaupels, F.; Geerlof, A.; Sarioglu, H.; Sattler, M.; Durner, J.; Lindermayr, C. Differential inhibition of Arabidopsis superoxide dismutases by peroxynitrite-mediated tyrosine nitration. J. Exp. Bot. 2015, 66, 989–999.
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619.
- Hervé-Grépinet, V.; Veillard, F.; Godat, E.; Heuzé-Vourc’h, N.; Lecaille, F.; Lalmanach, G. Extracellular catalase activity protects cysteine cathepsins from inactivation by hydrogen peroxide. EFEBS Lett. 2008, 582, 1307–1312.
- Lee, J.E.; Kim, H.; Jang, H.; Cho, E.J.; Youn, H.D. Hydrogen peroxide triggers the proteolytic cleavage and the inactivation of calcineurin. J. Neurochem. 2007, 100, 1703–1712.
- Yoon, S.O.; Park, S.J.; Yoon, S.Y.; Yun, C.H.; Chung, A.S. Sustained production of H(2)O(2) activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-kappa B pathway. J. Biol. Chem. 2002, 277, 30271–30282.
- Parakh, S.; Atkin, J.D. Novel roles for protein disulphide isomerase in disease states: A double edged sword? Front. Cell Dev. Biol. 2015, 3, 30.
- Bekendam, R.H.; Iyu, D.; Passam, F.; Stopa, J.D.; De Ceunynck, K.; Muse, O.; Bendapudi, P.K.; Garnier, C.L.; Gopal, S.; Crescence, L.; et al. Protein disulfide isomerase regulation by nitric oxide maintains vascular quiescence and controls thrombus formation. J. Thromb. Haemost. 2018, 16, 2322–2335.
- Nakamura, T.; Tu, S.; Akhtar, M.W.; Sunico, C.R.; Okamoto, S.; Lipton, S.A. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron 2013, 78, 596–614.
- Aranda, E.; Lopez-Pedrera, C.; De La Haba-Rodriguez, J.R.; Rodriguez-Ariza, A. Nitric oxide and cancer: The emerging role of S-nitrosylation. Curr. Mol. Med. 2012, 12, 50–67.
- Balkwill, F.R.; Capasso, M.; Hagemann, T. The tumor microenvironment at a glance. J. Cell Sci. 2012, 125 Pt 23, 5591–5596.
- Jezierska-Drutel, A.; Rosenzweig, S.A.; Neumann, C.A. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv. Cancer Res. 2013, 119, 107–125.
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773.
- Cook, J.A.; Gius, D.; Wink, D.A.; Krishna, M.C.; Russo, A.; Mitchell, J.B. Oxidative stress, redox, and the tumor microenvironment. Semin. Radiat. Oncol. 2004, 14, 259–266.
- Fiaschi, T.; Chiarugi, P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: A diabolic liaison. Int. J. Cell Biol. 2012, 2012, 762825.
- Ray, P.D.; Huang, B.W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990.
- Lorenzen, I.; Mullen, L.; Bekeschus, S.; Hanschmann, E.M. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxid. Med. Cell. Longev. 2017, 2017, 8459402.
- Noel, A.; Jost, M.; Maquoi, E. Matrix metalloproteinases at cancer tumor-host interface. Semin. Cell Dev. Biol. 2008, 19, 52–60.
- McCarthy, S.M.; Bove, P.F.; Matthews, D.E.; Akaike, T.; van der Vliet, A. Nitric oxide regulation of MMP-9 activation and its relationship to modifications of the cysteine switch. Biochemistry 2008, 47, 5832–5840.
- Reis, S.T.; Leite, K.R.M.; Piovesan, L.F.; Pontes-Junior, J.; Viana, N.I.; Abe, D.K.; Crippa, A.; Moura, C.M.; Adonias, S.P.; Srougi, M.; et al. Increased expression of MMP-9 and IL-8 are correlated with poor prognosis of Bladder Cancer. BMC Urol. 2012, 12, 18.
- Ferrerasa, M.; Felborb, U.; Lenharda, T.; Olsen, B.R.; Delaisse, J.M. Generation and degradation of human endostatin proteins by various proteinases. FEBS Lett. 2000, 486, 247–251.
- Harris, L.K.; McCormick, J.; Cartwright, J.E.; Whitley, G.S.; Dash, P.R. S-nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp. Cell Res. 2008, 314, 1765–1776.
- Eckert, R.L.; Kaartinen, M.T.; Nurminskaya, M.; Belkin, A.M.; Colak, G.; Johnson, G.V.; Mehta, K. Transglutaminase regulation of cell function. Physiol. Rev. 2014, 94, 383–417.
- Lai, T.S.; Hausladen, A.; Slaughter, T.F.; Eu, J.P.; Stamler, J.S.; Greenberg, C.S. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry 2001, 40, 4904–4910.
- Huang, L.; Xu, A.M.; Liu, W. Transglutaminase 2 in cancer. Am. J. Cancer Res. 2015, 5, 2756–2776.
- Mangala, L.S.; Mehta, K. Tissue transglutaminase (TG2) in cancer biology. Prog. Exp. Tumor Res. 2005, 38, 125–138.
- Fraser, M.; Chan, S.L.; Chan, S.S.; Fiscus, R.R.; Tsang, B.K. Regulation of p53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene 2006, 25, 2203–2212.
- Postovit, L.M. Role of Nitric Oxide in the Regulation of the Pro-tumourigenic Hypoxic Phenotype: From Instigation to Mitigation. In Nitric Oxide and Cancer: Pathogenesis and Therapy; Bonavida, B., Ed.; Springer Publishing: New York, NY, USA, 2015; pp. 65–84.
- Nath, N.; Kashfi, K.; Chen, J.; Rigas, B. Nitric oxide-donating aspirin inhibits β-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear β-catenin–TCF association. Proc. Natl. Acad. Sci. USA 2003, 100, 12584–12589.
- Dejam, A.; Hunter, C.J.; Pelletier, M.M.; Hsu, L.L.; Machado, R.F.; Shiva, S.; Power, G.G.; Kelm, M.; Gladwin, M.T.; Schechter, A.N. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood 2005, 106, 734–739.


