Nitric oxide (NO) is a highly reactive molecule, generated through metabolism of L-arginine by NO synthase (NOS). Abnormal NO levels in mammalian cells are associated with multiple human diseases, including cancer. Recent studies have uncovered that the NO signaling is compartmentalized, owing to the localization of NOS and the nature of biochemical reactions of NO, including S-nitrosylation. S-nitrosylation is a selective covalent post-translational modification adding a nitrosyl group to the reactive thiol group of a cysteine to form S-nitrosothiol (SNO), which is a key mechanism in transferring NO-mediated signals. While S-nitrosylation occurs only at select cysteine thiols, such a spatial constraint is partially resolved by transnitrosylation, where the nitrosyl moiety is transferred between two interacting proteins to successively transfer the NO signal to a distant location. As NOS is present in various subcellular locales, a stress could trigger concerted S-nitrosylation and transnitrosylation of a large number of proteins involved in divergent signaling cascades. S-nitrosylation is an emerging paradigm of redox signaling by which cells confer protection against oxidative stress.
- NO
- S-nitrosylation
- NOS
- ROS
- antioxidant
- redox regulation
Introduction
In the past decades, nitric oxide (NO) has garnered an increasing amount of interest with regard to its impact on many human diseases. Since its discovery by Furchgott et al. as “the endothelium-derived relaxing factor (EDRF)” over 30 years ago (1), NO has become one of the most studied subjects in the fields of biomedical sciences. NO was named “the Molecule of Year” in 1992, and its discovery led to the 1998 Nobel Prize of Furchgott et al. Despite the original conception of NO as a freely diffusible gas (2), NO production and signaling are rather compartmentalized and spatially regulated. This is owing to the facts that all the isoforms of NO synthases (NOS1-3) are mostly localized at the membrane and organelles (3-5), and that NO produced there is quickly (<0.1 sec) consumed by reacting with the proximal molecules (5-7). On one hand, NO reacts with molecular oxygen to become nitrogen oxide, which then reacts with a transition metal or cysteine thiol to form a nitrosyl adduct (metal nitrosylation or S-nitrosylation, respectively). The nitrosyl group could be further transferred to another protein in a distant site (transnitrosylation) to propagate NO signaling. Such effects of NO are said to be “direct” and involved in many anti-oxidant mechanisms as well as other biochemical activities (8). On the other hand, in the presence of high levels of reactive oxygen species (ROS), such as superoxide, NO reacts with ROS and forms a strong oxidant, reactive nitrogen species, which in turn reacts with and oxidatively damages a variety of biological molecules. This mode of NO effect is said to be “indirect” and involved in “pro-oxidant” signaling of NO (8).
The initial NO studies focused on the NO signaling pathway in specialized tissues such as neurons, muscles, endothelia and immune cells. For example, in the brain NO controls brain-blood flow and other processes that lead to normal brain function. In the circulatory system, NO controls leucocyte adhesion, pro-inflammatory signaling, platelet aggregation and angiogenesis (9). Such diverse functions of NO are mediated by the” classical” and “non-classical” schemes. In the classical NO signaling, NO binds the heme iron of guanylyl cyclase (sGC) to induce production of cGMP, which then activates the cGMP-dependent protein kinase (PKG) pathway (10). Notwithstanding this classical scheme, recent studies have unveiled a wealth of the “non-classical” NO signaling that mediates pleiotropic functions in diverse tissues/organs. A major mechanism of such new NO’s functions is S-nitrosylation, an NO-dependent covalent modification of a cysteine thiol (11). S-nitrosylation triggers a change in the protein structure, which could alter protein-protein interactions and enable further post-translational modifications such as phosphorylation, acetylation, ubiquitination and disulfide bond formation (11-14). Furthermore, in the presence of a low level of ROS, S-nitrosylation could not only scavenge NO to prevent it from reacting with ROS, but also protect cysteine thiols against ROS-mediated oxidation [sulfonic acid (RSO3H) formation] (15). At very high levels of ROS, however, NO could react with ROS to form a reactive nitrogen species as discussed above (8, 16).
S-nitrosylation could regulate >3000 proteins involved in a multitude of biological processes, including protein stability/turnover, steroid synthesis, transcription regulation, DNA damage repair, cellular growth/differentiation, apoptosis and redox regulation. As NOS could be present in various subcellular locales, a stress could trigger concerted S-nitrosylation and transnitrosylation of a large number of proteins involved in divergent signaling cascades. Dysregulated S-nitrosylation has been implicated in the incidence and progression of different diseases (11, 15, 17-19). In this review, we will provide the recent advances in our understanding of how S-nitrosylation exerts redox regulation on cells and tissues. We will especially focus on cellular locales which are equipped with various mechanisms that produce or fight against ROS, such as the mitochondrion, nucleus and extracellular environment. We will then discuss how dysregulated S-nitrosylation would lead to disease conditions.
NO and its roles in redox regulation
NO is a reactive, gaseous signaling molecule with a half-life of 0.09 ~ 2 seconds (6). NO is produced by nitric oxide synthase (NOS) using amino acid L-arginine as the substrate in a wide range of tissues and is involved in pleiotropic functions. Most NO’s studies have focused on its role in specialized cells, including neurons, muscles, endothelia and immune cells. In these cells, NO mediates neurotransmission, muscle and vessel dilation and pro-inflammatory signaling. Aside from these well-established roles, NO exerts pleiotropic functions in many different types of cells (20). These functions include regulation of tissue morphogenesis, polarity formation, cellular growth and movement (21-26).
Because of its nature as a radical, NO (•NO) is also involved in both anti- and pro-oxidant mechanisms. As an anti-oxidant, NO could modify cellular processes to confer protection to cells and tissues against oxidative damage (8). It is reported that pretreatment with NO could protect cells against the oxidative stress by hydrogen peroxide (H2O2) (27, 28), while NO could also limit the production of ROS by NADPH oxidase in immune cells (29). In addition, the beneficial effects of pretreatment with NO against ischemia–reperfusion-mediated tissue injury have been reported in clinical trials (30). Conversely, as a pro-oxidant, NO could react with molecular oxygen or ROS (especially, superoxide: O2-) and be converted to a strong oxidant, reactive nitrogen oxide species (RNOS, especially, peroxynitrite: ONOO−), that could oxidatively damage a variety of biological molecules.
It is proposed that whether NO exerts anti- or pro-oxidant effects depends on the concentration of NO (8). At lower NO concentrations, the activities of NO manifest through its direct interaction with the biological target, which are likely to lead to anti-oxidant effects. At higher NO concentrations, conversely, the activities of NO are indirectly mediated by RNOS derived from the reaction of NO with molecular oxygen or ROS, which are likely to lead to pro-oxidant effects. The deleterious effects of NO as a pro-oxidant often take place in confined locales where local NO concentrations could be quickly raised to higher levels (8). In fact, dose-dependent activities of NO have been reported by a number of previous studies. These studies suggest that the threshold concentration of NO that determines whether the effect is direct or indirect would be around 1 μM (8, 31).
Local NO concentration is in part controlled by spatial regulation and compartmentalization of the NO source. There are three isoforms of NOS (NOS1-3) that produce NO in mammalian cells. NOS-1 (neuronal NOS, nNOS) and NOS-3 (endothelial NOS, eNOS) are the constitutive NOS and are part of the membrane-bound protein complexes found in different subcellular locales. NO produced by NOS-1 or NOS-3 quickly (<0.1 sec) and directly reacts with the proximal targets, including NOS itself and the interacting proteins (3, 4, 6). In contrast, NOS-2 (iNOS) is the inducible NOS. In inflammatory cells, NOS-2 is initially expressed in the cytosol, but is recruited to phagosomes or peroxisomes to elevate the local NO concentration, where NO reacts with superoxide, forming peroxynitrite to kill pathogens (32, 33). Thus, dichotomous effects of NO as both an anti- and pro-oxidant are dependent on the source and location of NO that determines its local concentration.
NO production and biochemistry
Nitric Oxide Synthase (NOS)
The mammalian NOS consists of three isoforms (NOS1-3) that encompass ~50% homology (7). NOS-1 (nNOS) and -3 (eNOS) are expressed constitutively, while their activities are regulated post-translationally, such as by phosphorylation, S-nitrosylation, protein interaction, cofactor/substrate and calcium level. NOS-1 and NOS-3 produce a steady-state NO level to regulate tissue homeostasis and development (10, 34). Conversely, the expression of NOS-2 (iNOS) is regulated inducibly to produce a large amount of NO in response to an inflammatory signal (10, 34). In addition, mitochondria are reported to possess mtNOS (nNOS homologue) in the matrix and inner membrane. mtNOS is involved in the regulation of oxygen consumption and biogenesis of mitochondria (35-39). However, the actual presence of mtNOS is currently under debate (see below for details).
NOS is a dimer of two identical monomers tethered by tetrahedral coordination of a zinc ion at two CysXXXXCys motifs (each motif is contributed by a monomer). Through the motif, NOS binds the substrate L-arginine and cofactor tetrahydrobiopterin (BH4) which facilitates dimerization, substrate binding and enzymatic function (34, 40). The constitutive NOS-1 and -3, but not inducible NOS-2, are bound by calmodulin in response to increased calcium levels, which induces conformational changes to activate the enzymatic function (34, 41). Each NOS monomer is composed of two distinct domains: the carboxyl-terminal reductase and amino-terminal oxygenase domains. Both domains are connected by a linker region that binds calmodulin (34, 41). The reductase domain harbors a series of redox-active cofactors, NADPH, FAD and FMN, which serially transfer electrons. The oxygenase domain, serving as the dimerization site that coordinates a zinc ion, harbors another redox-active cofactor BH4, heme and the substrate L-arginine. In particular, BH4 plays an essential role in “coupling” the reductase and oxygenase domains (7, 34, 42, 43). It facilitates electron transfer from FMN of the reductase domain to the heme of the oxygenase domain. This causes the ferric (Fe3+) center of the heme to be reduced to the ferrous (Fe2+) center, which then reacts with molecular oxygen to form Fe2+-O2 complex. This complex, in turn, oxidizes the guanidine moiety of the substrate L-arginine, producing L-citrulline and NO (7, 34, 42, 43) (Figure 1, left).
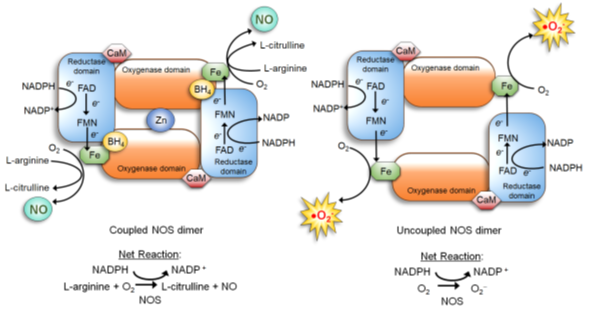
Figure 1. NOS dimer in the coupled (normal) vs. uncoupled states (40). (Left) In the normal coupled state, two NOS monomers are tethered by BH4, which also increases the binding affinity of the substrate L-arginine. Furthermore, the NOS dimer is stabilized by a zinc ion coordinated in the oxygenase domains of two monomers. Dimerization allows the coupling of two reactions: 1) electron flow: NADPHàFADàFMNà Fe of the hemeà molecular oxygen; and 2) oxidation of L-arginine. This coupling yields NO and L-citrulline. (Right) When BH4 level is scarce, NOS remains as a monomer and fails to bind L-arginine. The electron flow to molecular oxygen (Reaction 1) is uncoupled to L-arginine oxidation (Reaction 2), yielding superoxide (·O2-).
If the availability of the cofactor BH4 or the substrate L-arginine is reduced, however, NOS becomes “uncoupled” and unable to dimerize. The uncoupled NOS fails to transfer electrons from the reductase to the oxygenase domains and to oxidize L-arginine. Then, instead of producing NO, the heme of the oxygenase domain now produces superoxide (•O2-) (Figure 1, right), which further lowers the BH4 level by oxidizing it to biopterin (BH2) (43, 44). In fact, deficiency of BH4 is a major cause of NO deficiency in chronic disorders, such as diabetes (45), obesity (46), cardiovascular disease (40, 44), as well as cancer (47). NO deficiency could lead to tissue fibrosis and stiffening (48), which also increases cancer risks of patients (49).
BH4 deficiency could be attributed to its oxidative degradation to BH2 under hyperoxia or by a variety of biological oxidants, such as H2O2, peroxynitrite and heme (50, 51). On the other hand, BH4 deficiency could be ascribed to the deficiency of the enzymes that generate BH4 in the body. BH4 is either de novo synthesized from GTP or regenerated from the oxidized metabolites of BH4 (i.e., BH2 and sepiapterin) (52, 53). A defect of any enzyme involved in the two pathways could lead to BH4 deficiency. In particular, a mutation of GTP cyclohydrolase 1 (GCH1), the rate-limiting enzyme of BH4 biosynthesis, is a major genetic cause of BH4 deficiency found in different types of neurological and metabolic disorders (54, 55).
NO Signaling
NO signaling could be classified into the classical and non-classical schemes. In the classical scheme, NO binds to the heme group of soluble guanylyl cyclase (sGC) to promote the enzymatic activity that converts GTP to cGMP. cGMP serves as a second messenger and activates cGMP-dependent protein kinase (PKG). This lowers the levels of potassium and calcium ions in the cytosol, which hyperpolarizes the membrane potential and triggers neurotransmission (in neurons) and vasodilation (in vessels) (10, 34). Such classical NO pathway is said to be a “long-range” signaling, where the signal is passed through a relatively long distance from the NO sources. The examples of this pathway include paracrine/autocrine signaling of NO (56, 57).
On the other hand, the non-classical scheme of NO signaling includes covalent post-translational modifications of biomolecules by NO and NO derivatives. These are S-nitrosylation of protein thiols; metal nitrosylation of transition metals; and oxidative nitration or hydroxylation of various molecules (e.g., tyrosines, thiols, amines, fatty acids and guanine) (see below). This mode of NO pathway is said to be a “short-range” since the effect takes place in a relatively close range from the NO source and often within a certain subcellular localization (56). Nevertheless, recent evidence unveils that protein S-nitrosylation could be propagated beyond the boundary between different subcellular locales through transnitrosylation (see below) (58). S-nitrosylation is an emerging paradigm of redox signaling by which cells protect themselves against oxidative stress and reduce the levels of ROS (15, 59-62). We will focus on these antioxidant effects of protein S-nitrosylation in the following sections.
S-nitrosylation, Metal Nitrosylation and Nitration
Nitrosylation is the reaction to covalently incorporate the nitrosyl moiety of NO into another molecule. If nitrosylation takes place at the thiol group of a cysteine, this reaction is termed S-nitrosylation. On the other hand, if nitrosylation takes place at a transition metal (e.g., the catalytic site of a metalloenzyme), this reaction is termed metal nitrosylation.
S-nitrosylation takes place on the cysteine thiols at a physiological pH range (63). Although this reaction was discovered over 150 years ago (64), it has only been two decades since S-nitrosylation was recognized as an NO-mediated protein modification (65). Cumulative evidence has supported that S-nitrosylation is a ubiquitous regulatory mechanism for protein conformational change, protein-protein interactions and further post-translational modifications such as phosphorylation, acetylation, ubiquitination and disulfide bond formation (11-14, 58). S-nitrosylation regulates diverse processes, including transcription regulation, DNA damage repair, cellular growth/differentiation and apoptosis (11, 18, 19). Balanced regulation of S-nitrosylation is essential for normal pathophysiology, whereas its dysregulation leads to disease conditions (66). We will discuss the biological activities of S-nitrosylation in detail below.
For metal nitrosylation, NO interacts with metal centers of the heme, as first discovered by Keilin and Hartree 80 years ago (67). NO binding to the ferrous (Fe II) heme of sGC induces conformational change for the activation (68), whereas NO binding to the heme of cytochrome c oxidase in the mitochondrial electron transport chain causes inactivation (69). NO also binds to the ferrous hemoglobin with the higher affinity than those of oxygen and carbon oxide. In hyperoxic conditions such as ischemia reperfusion, NO binding to hemoglobin reduces the affinity of oxygen binding and confers protection to tissues against oxygen toxicity (70).
In the presence of high concentrations of NO and ROS, in particular superoxide, both molecules react to produce a highly destructive peroxynitrite, which could peroxidize lipids, nitrate tyrosines, thiols, amines and fatty acids, and hydroxylate guanine nucleotides at acidic pH (63). In particular, tyrosine-nitrated proteins are indicators of oxidative/nitrosative stress (63) and are degraded by 20S proteasome (71). Nitrated unsaturated fatty acids, on the other hand, form nitroalkenes or nitrohydroxyl derivatives that exert anti-inflammatory signals (72).
S-NITROSYLATION
Biochemistry of S-nitrosylation
NO itself is not an oxidant and does not strongly react with a protein thiol. Thus, for the majority of S-nitrosylation, NO first reacts with oxygen to increase the oxidation state and then reacts with a thiol (73). Different intermediate reactions have been proposed (56).
1) First, NO reacts with O2 to form a series of nitrogen oxides with increasing oxidation states (auto-oxidation). N2O3 reacts with a protein thiol to produce nitrite and a nitrosothiol. In particular, the rate of this reaction increases in hydrophobic environments such as membranes, the predominant locales of NOS1-3 (Figure 2, reaction 1) (56).
2) First, NO reacts with O2 to form NO2. NO2 reacts with a thiol to produce a thiol radical and nitrite. Then, NO reacts with a thiol radical to form a nitrosothiol (Figure 2, reaction 2) (56).
3) In the presence of a thiol radical, NO directly reacts with it to form a nitrosothiol (Figure 2, reaction 3) (56).
4) For a metal-catalyzed reaction, NO becomes oxidized by a transition metal, such as Fe3+ or Cu2+, in a metalloenzyme, forming nitrosonium (NO+). Nitrosonium then reacts with a thiol close to the catalytic center to form a nitrosothiol. This reaction takes place in specific cases, such as auto-nitrosylation of hemoglobin and GSNO formation by cytochrome c (Figure 2, reaction 4) (56).
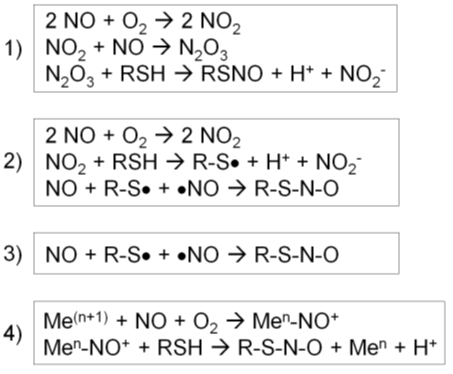
Figure 2. Four different types of S-nitrosylation reactions (56). 1) NO reacts with O2 to form a series of nitrogen oxides. N2O3 reacts with a protein thiol to produce nitrite and a nitrosothiol. 2) NO reacts with O2 to form NO2, which reacts with a thiol to produce a thiol radical and nitrite. Then, NO reacts with a thiol radical to form a nitrosothiol. 3) In the presence of a thiol radical, NO directly react with it to form a nitrosothiol. 4) NO becomes oxidized by a transition metal, forming nitrosonium (NO+). Nitrosonium then reacts with a thiol close to the catalytic center to form a nitrosothiol.
S-nitrosylation Reaction Specificity
Despite such abundance, S-nitrosylation occurs only at select cysteine residues of proteins. Such selectivity depends on the following criteria (5):
1) The target cysteines must be in the proximity of the NO source to increase the likelihood of S-nitrosylation (5).
- i) The first S-nitrosylation targets are the constitutive NOS (NOS-1 and NOS-3) themselves (3, 74). S-nitrosylation of NOS-3 inhibits its dimerization and thus activation (74), suggesting that S-nitrosylation of the constitutive NOS may serve as a self-shut-off mechanism. S-nitrosylation of the inducible NOS, NOS-2, on the other hand, is currently unknown.
- ii) The second S-nitrosylation targets are proteins directly interacting with NOS. NOS-1 interacts with a scaffolding protein DLG4 (a.k.a., PSD95) and a small G-protein Dexras1, which both become S-nitrosylated (3, 75). While the consequence of DLG4 S-nitrosylation is currently unknown, Dexras1 S-nitrosylation activates the protein function that promotes growth suppression and apoptosis of cells (75). NOS-3 interacts with the Hsp90 chaperon protein, which also becomes S-nitrosylated. Hsp90 S-nitrosylation inhibits its ATPase activity (4) and impairs the growth/survival of cancer cells dependent on Hsp90 chaperon functions (76). NOS-2 interacts with calcium- and zinc-binding proteins S100A8 and S100A9, which both become S-nitrosylated. S100A8/A9 are pro-inflammatory factors by their very nature. However, upon S-nitrosylation, they become converted to anti-inflammatory agents that inhibit mast cell activation and leukocyte-endothelium interaction (see the details below) (77-79).
iii) The third S-nitrosylation targets are proteins that are transnitrosylated by S-nitrosylated proteins (S-nitrosylases). For example, S100A8/A9, after being S-nitrosylated by NOS-2 (77, 78), transfer the nitrosyl group to their interacting proteins such as vimentin (78, 80, 81), conferring anti-inflammatory effects (79) (See below for more details).
2) The target cysteine must be within a signature motif, I/L-X-C-X2-D/E, which is specifically recognized by NOS (78). This motif must be within α-helix forming a large surface area to increase accessibility to reactants (78). Furthermore, within the S-nitrosylation motif, the target cysteine thiol must electrostatically interact with the neighboring charged residues (<6Å), which increases its nucleophilicity (i.e., reactivity) promoting its reactivity with the nitrosyl group (82, 83).
3) The target cysteine must be within a highly hydrophobic region formed by tertiary protein structure or membranes (11, 84). Because NO itself has low reduction potential (low tendency to receive electrons; oxidation state: +2), it cannot oxidize amino acids to form adducts at a physiological rate. Thus, NO-dependent amino acid oxidation is preceded by the oxidation of NO by molecular oxygen. This leads to formation of nitrogen oxide [NO2 (oxidation state: +4)] and N2O3 (Oxidation State: +3), which then elevates the reduction potential of the nitrosyl group by up to10 fold (85, 86). Because of such a critical role of NO’s reaction with molecular oxygen, S-nitrosylation of proteins preferentially takes place at cysteines in hydrophobic regions which could attract hydrophobic gases, NO and molecular oxygen, and enhance their reaction rate by 30-300 fold (84).
4) The target cysteine must be in the suitable environment. Which thiols are S-nitrosylated and what is the stability of the nitrosothiols, depend on the context and environmental factors (63). The accessibility of these target cysteines, which are often masked within hydrophobic regions of the protein, largely depends on the nature of neighboring residues. First, the pKa of the thiol is greatly influenced by the acidity and basicity of neighboring residues. Second, the presence of bulky amino acid residues (e.g., Phe, Tyr, Arg, and Leu) in the neighborhood elicits steric hindrance on the target cysteines. Thus, cysteines targeted for S-nitrosylation are those that have lower pKa, are surrounded by acidic and basic residues within 6 Å (promoting nucleophilicity of thiols), and are adjacent to few bulky residues (less sterically hindered) within 8 Å (87). Furthermore, at an acidic pH and in the presence of ROS, the same thiols could instead be targeted for nitration (oxidation) by NO as discussed above (63), which would then counteract their S-nitrosylation (15).
Other Factors that Control S-nitrosylation Level
The level of S-nitrosylation in the cell is also regulated by the following factors:
1) Redox status. Redox status modulates S-nitrosylation. High levels of antioxidants, which elevates the reducing potential of cells, could prevent S-nitrosylation, whereas a decrease of antioxidants promotes S-nitrosylation (61, 88, 89). For example, depletion of an antioxidant, glutathione (GSH), elevates S-nitrosylation of mitochondrial proteins (see below), which protects cells against thiol oxidation, permeabilization and apoptosis (90).
2) Denitrosylation. S-nitrosylation levels in cells are controlled by the balance between S-nitrosylation and denitrosylation. While S-nitrosylation is generally a non-enzymatic reaction (except for prokaryotes), denitrosylation could be a non-enzymatic or enzymatic reaction (91). Cleavage of S-nitrosyl group could spontaneously occur in the presence of reducing agents (ascorbate, glutathione), metal ions (Cu2+), heat, UV, ROS or nucleophiles (or anions) (91). In contrast, denitrosylation could be catalyzed by denitrosylases that enzymatically remove the S-nitrosyl group from cysteines. There are two major denitrosylase systems in cells: Thioredoxin (Trx)/thioredoxin reductase (TrxR) and S-nitrosoglutathione (GSNO)/GSNO reductase (GSNOR) systems. Trx/TrxR targets S-nitrosylated proteins that harbor C-X5-K or C-X6-K motif, including caspase-3 and nuclear factor-κB (NF-κB) (91). On the other hand, GSNOR is the alcohol dehydrogenase 5 (ADH5), and GSNO (the major endogenous S-nitrosyl donor, see below) is the only substrate of denitrosylation. GSNOR activity is inhibited by ROS, such as hydrogen peroxide, which elevates GSNO and antioxidant gene expression (92). The balance between S-nitrosylation and denitrosylation is essential for the normal physiological conditions. Overexpression or defect of GSNOR dysregulates S-nitrosylation levels and could trigger many diseases, including multi-organ dysfunction, sepsis and cancer (91). For example, the increase of GSNOR level in the aging brain contributes to cognitive impairment (93).
3) Endogenous NOS inhibitors. S-nitrosylation level could be regulated by NOS inhibitors that modulate NO levels. There are several endogenous NOS inhibitors which are classified into two categories: 1) L-arginine analogues (competitive inhibitors) and 2) allosteric NOS inhibitor.
1) L-arginine analogues: endogenous methylarginines (MAs): There are three endogenously produced MAs: asymmetric dimethylarginine (ADMA); symmetric dimethylarginine (SDMA) and NG-monomethyl-L-arginine (L-NMMA). They are formed by the liberation of methylated arginine residues from intracellular proteins. Out of the three MAs, ADMA and L-NMMA could bind the three isoforms of NOS at the L-arginine binding site to exert competitive inhibition. Both MAs are found in the blood, where ADMA is 5-10 times more abundant than L-NMMA (94). Especially, ADMA levels are elevated in patients with different diseases, cardiovascular disease, diabetes, Alzheimer’s disease, liver failure and kidney failure (94).
2) Allosteric NOS inhibitor: Dynein light chain LC8-Type 1 (DYNLL1, PIN) is the 8 kDa cross-linker of dynein to cargos and to adapter proteins. DYNLL1 binds to the N-terminal PDZ domain of NOS1 (absent in NOS2 and NOS3) and inhibits its dimerization, inactivating the enzymatic function. DYLL1 level quickly increases in response to ischemia, intended to counteract the rise in NOS1 activity to protect the tissue against damage by excessive NO (95).
TRANSNITROSYLATION
Reaction
The selectivity of S-nitrosylation sites may be ameliorated by successive S-nitrosylation reactions termed transnitrosylation (58). Transnitrosylation is a process where a protein (nitrosylase), which is S-nitrosylated at cysteine(s) or nitrosylated at the metal center (e.g., heme), interacts with a protein containing the I/L-X-C-X2-D/E motif, leading to the transfer of the nitrosyl moiety to the interacting cysteine thiol. Such nitrosylation may take place successively, allowing for signal transmission to a site distant from the NO source. These reactions take place in two scenarios: Cys-to-Cys or Metal-to-Cys transnitrosylation (Figure 3) (58, 78, 85).
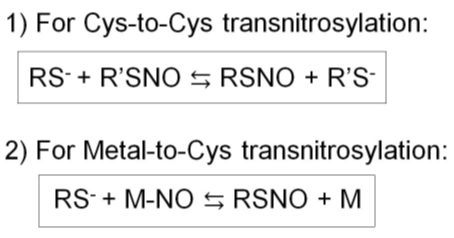
Figure 3. Two types of transnitrosylation reactions. 1) Cys-to-Cys transnitrosylation. 2) Metal-to-Cys transnitrosylation.
In the metal-mediated transnitrosylation, the nitrosyl group may be transferred intramolecularly or intermolecularly. In hemoglobin, NO is transferred from the heme iron to the neighboring cysteine thiols in the same molecule, whereas in cytochrome c, the metal-coordinated NO is transferred to the thiols of an interacting glutathione leading to the formation of GSNO (See below) (66, 96).
Transnitrosylation involves the nucleophilic attack of the recipient’s thiolate anion on the nitrosyl nitrogen of the donor (nitrosylase) (85). So far, less than 10 S-nitrosylases have been identified, and only specific cysteines are targeted for S-nitrosylation. This enables selective activation/inhibition of particular signaling pathways (58). A major determinant for the selective NO transfer is the physical distance between the donor (S-nitrosylase) and recipient thiol groups of cysteines. Another determinant is the redox potential between the donor and recipient thiols (58). Thus, transnitrosylation occurs only when two proteins directly interact and possess appropriate redox potentials allowing for electron transfer followed by NO transfer (58). It is speculated that physical association of the two proteins triggers conformational change, allowing the recipient thiol to form thiolate anion (RS−) which then attacks the nitrosyl group of the donor (58). There are fewer than 10 transnitrosylation reactions known up to date (58). Among these reactions, some of the major signaling pathways are listed below.
S-nitrosylases or Transnitrosylases
S-nitrosoglutathione (GSNO)
S-nitrosoglutathione (GSNO) is the most abundant S-nitrosothiol and the major endogenous NO donor for proteins everywhere in the cell. GSNO is generated in the mitochondria when the nitrosyl group is transferred from the heme iron of cytochrome c to glutathione (GSH) (96). GSNO then translocates to different subcellular locales and transnitrosylates the interacting proteins including NF-κB, STAT3, AKT, EGFR and IGF-1R (97-99). GSNO-mediated transnitrosylation of the p65 and p50 subunits of NF-κB, as well as IκB kinase, inhibits NF-κB activation that is closely associated with malignant behavior of cancers (100-102). One of the NF-κB-mediated oncogenic signaling is to upregulate IL6, which then activates STAT3, a transcription factor that promotes cell survival and proliferation (103). STAT3, downstream of NF-κB, is also targeted for transnitrosylation by GSNO, which inhibits STAT3 phosphorylation required for its activation (101). Additionally, cell surface receptors and the associated proteins, such as AKT, EGFR and IGF-1R, are transnitrosylated by GSNO, leading to inhibition of their phosphorylation-dependent activation (99). Accordingly, preclinical studies have reported the potent anti-cancer effects of GSNO in suppressing tumor cell growth and improving the efficacy of radiation therapy (98, 101).
Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH)
Another important S-nitrosylase is the glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH). GAPDH is well known for its role in catalyzing the conversion of glyceraldehyde 3-phosphate to D-glycerate 1,3-bisphosphate in the cytosolic glycolysis (104). GAPDH also plays critical roles as a transnitrosylase in the nucleus and mitochondria, including transcriptional regulation and apoptosis (105).
GAPDH translocates from the cytosol to the nucleus upon S-nitrosylation at the catalytic Cys150 by S100A8/A9 that has been S-nitrosylated by NOS-2 in response to a stress (78, 106). S-nitrosylation of GAPDH enables its interaction with an E3 ubiquitin ligase Siah1 which has the nuclear localization signal (NLS). GAPDH-Siah1 complex then translocates to the nucleus, where Siah1 mediates ubiquitination and degradation of nuclear proteins to initiate apoptosis. In contrast, GAPDH forms a complex with p53 to activate p53-mediated apoptosis (105, 107-109). Furthermore, GAPDH transnitrosylates proteins involved in transcription and DNA repair. These proteins include deacetylases: sirtuin-1 (SIRT1) and HDAC2, inhibited by S-nitrosylation; and DNA repair protein: DNA-activating protein kinase (DNA-PK), activated by S-nitrosylation (105, 106).
In response to a stress, GAPDH also translocates to mitochondria to transnitrosylate mitochondrial proteins, such as Hsp60, acetyl-CoA acetyltransferase (ACAT1) and voltage-dependent anion channel 1 (VDAC1) (110). The elevation of GAPDH-mediated transnitrosylation of mitochondrial proteins regulates the permeability of the mitochondrial membrane, mitochondrial functions and cell death (111).
In cancer cells, GAPDH-mediated transnitrosylation of nuclear proteins is impaired, leading to a defect of stress-responsive apoptosis (106, 108, 112, 113). One reason for this impairment is the downregulation of Siah1 that facilitates the nuclear translocation of GAPDH (112). Siah1 expression is directly regulated by p53, the tumor suppressor downregulated in many cancer types (112, 114).
S100A8/S100A9
The third example of S-nitrosylases are S100A8 and S100A9, which are calcium- and zinc-binding proteins playing roles in the regulation of inflammatory processes and immune response, such as neutrophil chemotaxis and adhesion (77). They represent up to 40 % of neutrophil cytoplasmic proteins, but are secreted in response to the elevated level of ROS at the site of inflammation (77, 115). S100A8 and S100A9 predominantly exist as calprotectin (S100A8/A9 heterodimer) which forms a complex with arachidonic acid (AA) released upon tissue injury. S100A8/A9-AA complex interacts with NADPH oxidase (NOX), and AA is transferred to NOX to activate its catalytic activity to produce ROS (see below)(116).
However, S100A8/A9 are both targeted for S-nitrosylation and converted to anti-inflammatory agents that inhibit mast cell activation and leukocyte-endothelium interaction (79, 115). Furthermore, S-nitrosylated S100A8/A9 transnitrosylates their interacting proteins to transmit anti-inflammatory signals to distant sites. Upon NOS-2 induction under inflammatory stimuli, S100A8/A9 assists in the formation of a linkage between NOS-2 and the target proteins, allowing the nitrosyl group to be transferred from NOS-2 to the target (77, 78). Over 100 proteins transnitrosylated by S100A8/A9 have been so far identified within cells and in microcirculation (78). These targets include GAPDH and hemoglobin (see above). S100A8/A9 also transnitrosylates some cytoskeletal elements such as ERM proteins (ezrin and moesin), linking cortical actin to the plasma membrane, and vimentin, the major intermediate filament in mesenchymal cells and metastatic cancer cells (78-81). The direct consequence of S-nitrosylation of these cytoskeletal elements is yet to be determined. Nevertheless, it is proposed that S-nitrosylation of these proteins induces conformational changes, influencing protein stability and protein-protein interactions (117, 118).
S-NITROSYLATION OF CELLULAR PROTEINS FOR REDOX REGULATION
Inside the cell, S-nitrosylation levels of cytosolic proteins are relatively low. One reason for this is that the S-nitrosyl group could be spontaneously cleaved by various factors in the cytosol. These factors include reducing agents (ascorbate and glutathione), metal ions (Cu2+), heat, UV, ROS and nucleophiles (or anions) (17, 91). The other reason is that the subcellular localizations of the constitutive NOS-1 and NO-2 are compartmentalized, which also compartmentalizes the majority of S-nitrosylation reactions (3, 4, 6, 117). Although NOS-2 is initially expressed in the cytosol, NO produced by NOS-2 is more likely to react with ROS to form RNOS than S-nitrosylating protein thiols (32, 33). Thus, the major sites of S-nitrosylation are organelles, (e.g., mitochondria, Golgi apparatus, and endoplasmic reticulum), nucleus and plasma membrane. Among these sites, we will feature mitochondria and nuclei where S-nitrosylation of proteins plays critical roles in protection of cells against oxidative stress. Some of these proteins are listed in Table 1.
Table 1. Effect of S-nitrosylation of proteins associated with Tumor Microenvironment.
|
Protein Name |
Effect of S-Nitrosylation |
Consequence |
Ref. |
|
Bcl-2 |
Inhibition of ubiquitination (activation) |
Anti-apoptosis |
(18) |
|
Caspases |
Inhibition of caspase activity |
Anti-apoptosis |
(119, 120) |
|
Fas Receptor |
Promote Fas ligand mediated apoptosis |
Apoptosis |
(18) |
|
GAPDH |
Nuclear translocation |
Apoptosis |
(121) |
|
TG2 |
Inhibition of activity |
Tumor suppression |
(122, 123) |
|
MGMT |
Inhibition of DNA repair activity |
DNA damage & apoptosis |
(18, 124, 125) |
|
OGG1 |
Inhibition of DNA repair activity |
DNA damage & apoptosis |
(124) |
|
HDAC2 |
Inhibition of deacetylase activity |
Histone acetylation |
(126, 127) |
|
MMPs |
Activation (low level) Inhibition (high level) |
Tumor progression Tumor suppression |
(128, 129) |
|
Caveolin-1 |
Inhibition of proteasomal degradation |
Tumor progression |
(130) |
|
c-Src |
Activation |
Tumor progression |
(18) |
|
Β-catenin |
Proteasomal degradation |
Tumor suppression |
(131) |
|
HDM2 |
Inhibition of binding to p53 |
Tumor suppression |
(132) |
|
HIF-1α |
Stabilization of activity |
Pro-angiogenesis |
(18, 133) |
|
MAT |
Inactivation of enzyme activity |
Tumor suppression |
(134, 135) |
|
NF-kB |
Inhibition of activity |
Tumor suppression |
(133) |
|
PTEN |
Inhibition of activity |
Tumor progression |
(18, 136) |
|
Ras |
Activation |
Tumor progression |
(137) |
S-nitrosylation of Redox-sensitive Mitochondrial Proteins
Mitochondria play key roles in the production of ROS as well as conferring antioxidant mechanisms along with all other functions (Figure 4) (138). The bioactivities and quality control of mitochondria are in part regulated by S-nitrosylation and denitrosylation of mitochondrial proteins (139, 140). Mitochondrial proteins become S-nitrosylated in response to changes in mitochondrial respiration and redox equilibrium. This results in the protection of protein thiols and the cell against oxidative damage and death, while also preventing further ROS production (59-61). Such responsiveness of S-nitrosylation to the oxidizing environment could be in part ascribed to the nature of its biochemistry. NO itself is not an oxidant and would need to react with molecular oxygen to increase the oxidation state in order to react with a protein thiol (73).
The source of mitochondrial NO remains under debate. On one hand, mitochondrial NO may be generated by mitochondrial NOS (mtNOS) (60). mtNOS is reported to be calcium-sensitive and constitutively active (homologous to NOS-1 [nNOS]) and integral to the inner mitochondrial membrane (36). However, the existence of mtNOS remains controversial (60). On the other hand, mitochondrial NO may be derived from NO generated in the cytoplasm and transported via a transnitrosylase through a cognate transporter on the mitochondrial membranes (61, 141).
NO produced in the mitochondria could S-nitrosylate or Fe-nitrosylate proteins preferentially in the inner mitochondrial membrane and intermembrane space because of their lower pH and lipophilic environments. Mitochondrial proteins targeted for S-nitrosylation include all the complexes in the electron transport chain (ETC): Complex I (NADH: ubiquinone oxidoreductase); Complex II (succinate dehydrogenase); Complex III (cytochrome b-c1 Complex); cytochrome c, Complex IV (cytochrome C oxidase) as well as ATP synthase (Complex V) (59, 60). In addition, several metabolic enzymes involved in citric acid (Krebs) cycle and β-oxidation (60, 142), as well as different mitochondrial proteins involved in regulation of apoptosis (143-145), are S-nitrosylated. Such S-nitrosylation of mitochondrial proteins mostly inhibit their activities to regulate mitochondrial O2 consumption, ROS production and mitophagy, while protecting the cell against death signals (Figure 4) (36).
On the other hand, in the presence of large amounts of ROS (and/or NO), NO could react with superoxide to produce a powerful oxidant, peroxynitrite (ONOO(-)) that can irreversibly nitrate or peroxidate tyrosines and unsaturated fatty acids (141, 146).
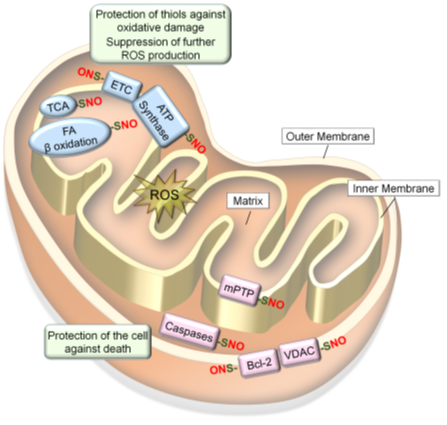
Figure 4. S-nitrosylation of mitochondrial proteins confer resistance to oxidative stress. Mitochondrial proteins become S-nitrosylated in oxidizing environments. S-nitrosylation of proteins involved in respiration and energy production (ETC, ATP synthesis, TCA cycle and fatty acid β oxidation) leads to prevention of ROS production and protection of protein thiols in these enzymes against oxidative damage. In contrast, S-nitrosylation of proteins involved in cell death regulation (mPTP, caspases-3/-9, VDAC and Bcl-2) leads to protection of the cell against death signals.
Electron Transport Chain (ETC) and ATP Synthase.
The electron transport chain (ETC, respiratory chain) is composed of Complexes I-IV bound to the inner membrane of the mitochondria. Electrons are sequentially passed from Complex I to IV through a series of redox reactions, which releases energy to generate proton gradient across the inner membrane. Proton gradient then triggers chemiosmosis to drive ATP synthesis (Complex V). The entire process, termed oxidative phosphorylation, is the major source of ATP in aerobic organisms (147). On the other hand, ETC is the primary site of ROS production in the cell. ROS production becomes the most prominent when ETC and ATP synthesis are uncoupled due to the insufficient proton gradient across the inner membrane. Such uncoupling causes reverse electron transport (RET) at Complex I, leading to production of large amounts of superoxide (148). The amount of ROS generated may often exceed the amount catalyzed by mitochondrial antioxidant enzymes, superoxide dismutase (SDS) and catalase (CAT).
In oxidizing conditions, the enzymatic activities of Complexes I-V could be inhibited by S-nitrosylation or Fe-nitrosylation, preventing further ROS production (59-61). For example, O2 binding to the Fe-heme center of Complex IV could be competitively inhibited by NO binding, which reduces oxygen consumption and thus further production of ROS (149). In addition, S-nitrosylation of Complexes not only protects themselves against oxidative damage by ROS, but also scavenges NO to prevent it from reacting with superoxide to form peroxynitrite (146). Furthermore, in hypoxic conditions, nitric oxide production becomes elevated (150), which could also promote S-nitrosylation and inactivation of mitochondrial ETC proteins (149). This reduces mitochondrial oxygen consumption and allows for the redistribution of intracellular oxygen to other locales. This would also prevent the stabilization of hypoxia-inducible factor-1α (HIF-1α) in response to hypoxia (151).
Inactivation of Complexes 1-V ultimately impedes oxidative phosphorylation (i.e., cellular respiration), which disrupts protein pumping and causes mitochondrial depolarization (39, 59). This activates the PINK1/Parkin pathway that induces mitophagy to selectively eliminate dysfunctional mitochondria (152). Mitophagy is a mechanism by which mitochondria protect the cell against oxidative stress (153).
In addition to the five Complexes, cytochrome c, which carries electrons between Complexes III and IV, is also targeted for S-nitrosylation. Cytochrome c becomes nitrosylated at the iron of the heme moiety, which then transfers the nitrosyl group to glutathione (GSH), generating the abundant transnitrosylase, GSNO (154). Although S-nitrosylated cytochrome c is released to the cytosol during apoptosis (155), it retains its ability to synthesize GSNO and is proposed to exert suppression on apoptotic signaling (154).
Metabolic enzymes.
Mitochondrial inner membrane and matrix are the sites of aerobic oxidation of pyruvate (citric acid [Krebs, TCA] cycle) and fatty acids (fatty acid β-oxidation) to CO2. These catabolic reactions could not only generate ROS by themselves (156-158), but also serve as the sources of electrons and co-enzymes (e.g., NADH and FADH2) to drive the ETC that produces ROS (157, 159). Under oxidizing conditions, many of these catabolic enzymes are targeted for S-nitrosylation to inhibit their activities (59, 60, 142, 160). These enzymes include three from citric acid (Krebs) cycle (aconitase; α-ketoglutarate dehydrogenase; and the succinate dehydrogenase [Complex II]); and five from fatty acid β-oxidation (long-chain and short-chain acyl-CoA dehydrogenase; enoyl-CoA hydratase; carnitine palmitoyl transferase 2 and flavoprotein dehydrogenase) (60). Inhibition of these catabolic enzymes contributes to the reduction of ROS production.
Mitochondrial pro- and anti-apoptotic proteins.
As described above, S-nitrosylation of the ETC and mitochondrial metabolic enzymes in the oxidizing environment prevents further production of ROS and protects protein thiols against oxidative damage. On the other hand, S-nitrosylation of pro- and anti-apoptotic/necrotic mitochondrial proteins protects the cell against death. The reason for such dichotomous effects could be that because reduced energy production (by inhibition of the ETC and metabolic enzymes) would otherwise induce cell death, death pathways are also inhibited for counteraction. These pro- and anti-apoptotic proteins include mitochondrial permeability transition pore (mPTP), voltage-dependent anion channel (VDAC), caspases and Bcl-2
mPTP is formed in the inner mitochondrial membrane under a stress (e.g., oxidative stress) and plays a pivotal role in controlling the mitochondrial membrane potential. Opening of the pore under stress increases the permeability of the mitochondrial membrane to molecules <1.5 kDa, leading to mitochondrial swelling and necrosis (161). NO produced at the basal level (e.g., 5 μM) could S-nitrosylate cyclophilin D (CypD), a critical mPTP regulatory component. This prevents the association of CypD with mPTP that is required for opening the pore (143, 162) and confers a protection to the cell under a stress. On the other hand, NO produced at a high concentration (e.g., 500 μM) could produce peroxynitrite in the presence of large amounts of ROS. Peroxynitrite could oxidize mPTP to induce multiple disulfide-linkages, which would lead to the opening of mPTP, loss of ATP production and necrosis (163).
VDAC is located in the mitochondrial outer membrane and controls the influx and efflux of mitochondrial metabolites. In addition, VDAC plays a major role in mitochondrial-mediated apoptosis through regulation of the release of cytochrome c that activates caspase-9 for apoptotic signaling (164). The effect of S-nitrosylation on VDAC is reported to be biphasic, similar to mPTP. Low levels (< 1 μM) of NO (and S-nitrosylation) inhibit VDAC function, conferring protection to the cell against apoptotic activation. On the other hand, higher levels of NO upregulate the function (165), although it is unknown whether this also involves the production of peroxynitrite or other mechanisms.
Caspases are the major executors of apoptosis located in the cytoplasm and mitochondrial intermembrane space. In the absence of apoptotic signal, zymogens of caspase-3 and caspase-9 in mitochondria are S-nitrosylated at the catalytic sides to prevent their activities. Upon activation of Fas receptor, zymogens become denitrosylated allowing for their further activation (144).
Bcl-2 family proteins are localized on the outer mitochondrial membrane and regulate apoptosis by controlling the release of pro-apoptotic intermembrane proteins (e.g., cytochrome c) (166). The anti-apoptotic Bcl-2 becomes S-nitrosylated in response to apoptotic stimuli. This inhibits ubiquitine-proteasomal degradation of Bcl-2 and confers protection to the cell against apoptosis (145).
S-nitrosylation of Redox-regulatory Nuclear Proteins
A number of nuclear proteins are targeted for S-nitrosylation in response to a stress, in particular oxidative stress, leading to up- or down-modulation of the activities. For the potential source of the nitrosyl group in the nucleus, two possibilities have been proposed: one is by NOS translocated to the nucleus; and the other is by transnitrosylases localized in the nucleus. NOS 1-3 have been shown to be translocated to the nucleus in response to a stimulus, such as a mitogen, oxidative stress and other pathological conditions. In the nucleus, NOS isoforms regulate S-nitrosylation of nuclear proteins, gene expression and calcium homeostasis (167-170). On the other hand, a transnitrosylase, GAPDH, becomes S-nitrosylated in the cytosol in response to a stress (such as oxidative stress) and translocates to the nucleus, where it transnitrosylates a number of nuclear proteins, including transcription factors, nuclear translocators and DNA repair proteins, involved in redox regulation and protection of the cell against stress.
Transcription Factors.
Several redox-regulatory transcription factors are S-nitrosylated in response to a change in redox homeostasis, which alters the expression of their target genes to protect the cell against oxidative stress. These transcription factors include nuclear factor (erythroid-derived 2)-like 2 (NRF2); p53, nuclear factor κB (NF-κB); and hypoxia-inducible factor-1α (HIF-1α).
NRF2 is the master regulator of the transcription of antioxidant genes. In response to oxidative stress, NRF2 binds a vast range of gene promoters that harbor the antioxidant-response elements (ARE). These genes include enzymes involved in glutathione and thioredoxin systems (e.g., glutamate-cysteine ligase catalytic subunit [GCLC]), mitochondrial superoxide dismutase (Mn2+ SOD, SOD2), peroxiredoxin, HSP70 and ferritin (171, 172). NRF2 is sequestered in the cytoplasm by formation of a latent complex with Klech-like ECH-associated protein 1 (Keap1), whereas dissociation from Keap1 enables nuclear translocation and activation NRF2 (173). Among all other stimuli, S-nitrosylation of Keap1 is one of the major mechanisms that induce NRF2 activation (174).
p53 tumor suppressor protein is another transcription factor involved in redox regulation. In response to a mild oxidative stress, p53 becomes S-nitrosylated at Cys124 in the DNA binding domain by the nuclear-localized nNOS. p53 then binds and transactivates the target genes, such as peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), a powerful controller of antioxidant mechanisms. PGC-1α interacts with NRF2 and serves as the co-activator of NRF2-mediated expression of a host of antioxidant genes (e.g., SOD2 and GCLC, see above) (175-177).S-nitrosylation also takes place in MDM2, an E3 ubiquitin protein ligase that binds and targets p53 for degradation. S-nitrosylation of MDM2 inhibits its interaction with p53, thus stabilizing p53 (178). Although Cys124 of p53 is not a mutation hotspot, nuclear S-nitrosylation of p53 could decline as the nuclear localized nNOS decreases during aging (176).
NF-κB is a transcription factor that regulates the expression of genes critically involved in inflammation and cell proliferation. NF-κB elevates mitochondrial ETC and ROS production, while counteracting the NRF2 pathway (179). NF-κB is usually bounded by the inhibitor I- κB and sequestered in the cytoplasm. However, in response to a stress such as ROS, I-κB becomes phosphorylated by I-κB-kinase complex (IKK- α, - β, and -γ), which leads to ubiquitine-proteasome-mediated degradation of I-κB and nuclear translocation and activation NF-κB (180). IKK- β could be inhibited by S-nitrosylation of the catalytic site, which prevents activation of NF-κB (181) and contributes to the reduction of mitochondrial ROS production (182).
HIF-1 is a transcription regulator that is regulated by oxygen levels. HIF-1 is a heterodimer composed of subunits HIF-1α and HIF-1β. Under normoxic condition, HIF-1α is hydroxylated at proline and arginine residues, which leads to its degradation mediated by the ubiquitine-proteasome system. In response to hypoxia, hydroxylation is inhibited, and the stable HIF-1 dimer translocates to the nucleus, where it transactivates hypoxia-responsive genes, such as vascular endothelial growth factor (VEGF) that promotes angiogenesis (183). However, during hypoxia, NO could help suppress HIF-1 activation by inhibiting mitochondrial ETC via S-nitrosylation, which then increases the oxygen availability in the intercellular compartments outside mitochondria (14). On the other hand, in normoxia, NO could S-nitrosylate HIF-1α, which helps stabilize HIF-1α and activate HIF-1-mediated expression of the target genes (184), demonstrating the context-dependence of NO-mediated HIF-1 regulation.
Nuclear Translocator.
The activities of signaling proteins and transcription factors are often regulated by their translocation between the nucleus and cytosol. This process is facilitated by the karyopherin family members that recognize the nuclear localization signal (NLS) or nuclear export signal (NES) of the cargos and translocate them through the nuclear pore complex (185). One of these transporters is chromosomal region maintenance 1 (CRM1; a.k.a., Exportin-1), which is involved in redox-regulation.
CRM1 facilitates nuclear export of over 200 nuclear proteins that possess the leucin-rich nuclear export signals (NESs) and play pivotal roles in homeostasis and stress response of cells. These cargo proteins include NRF2, p53, p73, MDM2, BRCA1/2, SMAD1/4, STAT1, IκB, c-Abl and FOXO-3A (186-189). These cargo proteins are exported from the nucleus by CRM1 and sequestered or targeted for degradation in the cytosol (189, 190). CRM1 activity is inhibited by S-nitrosylation upon an increase of the nitrosyl group in the nucleus. S-nitrosylation compromises the ability of CRM1 to recognize and bind NESs (189). One of the major results of such CRM1 inhibition is the nuclear accumulation of NRF2 and induction of the antioxidant defense mechanism (see above) (189).
DNA Damage Repair Proteins.
Some DNA damage repair proteins in the nucleus interact with and become transnitrosylated by S-nitrosylated GAPDH. These proteins include the catalytic subunit of DNA-activated protein kinase (DNA-PKcs) (106, 191) and Apurinic-apyrimidinic (AP) endonuclease 1 (APE1)(192).
DNA-PKcs plays critical roles in the repair of the damaged DNA through non-homologous end joining (193). DNA-PKcs interacts with and become transnitrosylated by GAPDH which has translocated to the nucleus upon S-nitrosylation (78, 106). Transnitrosylation of DNA-PKcs upregulates its DNA repair activity in response to DNA-damaging anti-tumor agents (194, 195).
APE1 (also known as redox factor 1, Ref-1) is involved in the base excision repair of damaged DNA as well as regulation of cellular redox response (196). Although APE1 is predominantly localized in the nucleus, its subcellular localization is dynamically regulated and it could translocate to the mitochondria and cytoplasm (197). APE1 interacts with GAPDH which has translocated to the nucleus upon S-nitrosylation (78, 106). APE1 interaction with GAPDH is not only critical for its nuclease activity (192), but also induces transnitrosylation of APE1. Transnitrosylation of APE1 triggers its nuclear export, independent of the nuclear exporter CRM1 (see above), allowing for its translocation to different subcellular locales (198).
S-NITROSYLATION OF EXTRACELLULAR PROTEINS FOR REDOX REGULATORS
The extracellular environment is oxidizing, characterized by the presence of a vast array of proteins linked via disulfide bonds. This is in stark contrast with the intracellular environment that has the highly reducing nature, limiting the formation of disulfide bonds (199). ROS in the extracellular environment could be derived from that produced in the intracellular environment which passes through a specific transporter [e.g., aquaporin 1 [see below]). However, in certain types of cells (e.g., immune cells and vascular cells), NADPH oxidase (NOX) is the major source of the extracellular ROS, producing and secreting ROS into the extracellular environment and circulation (Figure 5)(199). In addition, protein disulfide isomerase (PDI) complexes with NOX to promote the production of the extracellular ROS. To counteract the extracellular ROS, cells secrete different antioxidant enzymes, including extracellular superoxide dismutase (Ec-SOD, SOD3) which catalyzes the dismutation of superoxide to hydrogen peroxide (199), catalase (CAT), glutathione peroxidase (GPx) and thioredoxin (Trx), which remove hydrogen peroxide (200-202). All these proteins are targeted for S-nitrosylation to regulate their activities.
Sources of the Extracellular NO and Nitrosyl group
Cumulative evidence unveils that a number of extracellular proteins are S-nitrosylated (203). This has led to the active discussion of the possible sources of the extracellular nitrosyl group (Figure 5). There have been at least three mechanisms proposed: One possible mechanism is that NO passes through the membrane by itself. It has been reported that NO could freely diffuse through the phospholipid bilayer as efficiently as molecular oxygen (204). In addition, NO could be actively transported through a water channel protein aquaporin-1 (AQP1) (205). Such NO efflux is proposed to play pivotal roles in regulating NO’s functions in diverse tissues, including immune cells, neurons and vasculature (205-207). The second possible mechanism is that the nitrosyl group is transferred from a major transnitrosylase, GSNO, to a free amino acid cysteine. Then, S-nitrosylated cysteine passes through the cognate amino acid transporter and enters the extracellular space (208). The third possible mechanism is that secretory proteins become S-nitrosylated prior to secretion. Secretory proteins are processed in the Golgi apparatus prior to exocytosis. It is reported that many proteins found in the Golgi are S-nitrosylated by the Golgi-localized NOS3 (209, 210).
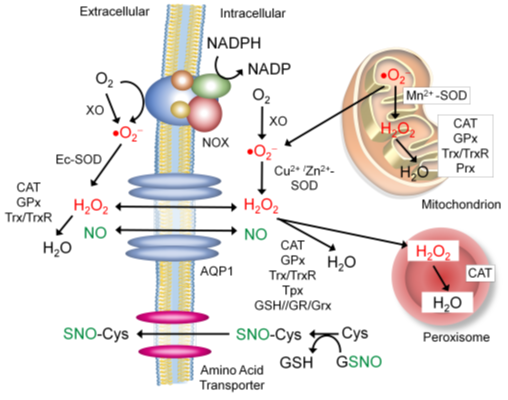
Figure 5. Trafficking of the extracellular and intracellular ROS and NO. Abbreviations: CAT: catalase, Cu2+/Zn2+-SOD: cupper/zinc superoxide manganese, Ec-SOD: extracellular superoxide dismutase, SNO: S-nitrosocysteine, GPx: glutathione peroxidase, GSH: glutathione, GSNO: S-nitrothiolglutathione, GR: glutathione reductase, Grx: glutaredoxin, Mn2+-SOD: manganese superoxide dismutase, NOX: NADPH oxidase, Prx: peroxiredoxin, Trx: thioredoxin, TrxR: thioredoxin reductase, and XO: xanthine oxidase.
Proteins that Regulate Extracellular ROS and NO Levels
Aquaporin-1 (APQ-1).
APQ-1 is a membrane-bound water channel protein expressed in various types of tissues. It transports water as well as low molecular weight gases such as NO and hydrogen peroxide between the intracellular and extracellular spaces (205, 211). APQ-1 is targeted for S-nitrosylation at the cysteine residue within the functional pore (212, 213). S-nitrosylation is mediated by NO produced from NOS3, co-localized with APQ1 within caveolae. This leads to the inhibition of the channel function of APQ-1, suggesting a possible negative feedback regulation by NO (200).
NADPH Oxidase (NOX).
NOX is a membrane-bound enzyme complex containing the catalytic heme moiety. NOX is localized on the plasma membrane as well as on the membrane of phagosomes of vascular cells and immune cells. NOX complex faces the extracellular space and produces superoxide or hydrogen peroxide into the extracellular environment by transferring electrons from NADPH to molecular oxygen bound to a heme (214). NOX is activated by binding arachidonic acid (AA), transferred from S100A8/A9-AA complex (see above)(116). AA binding facilitates the assembly of the NOX enzyme complex (215). In disease conditions, NOX2 and NOX4 expression could be elevated by TGFβ signaling, which could exacerbate oxidative tissue injury (216). On the other hand, NOX enzymatic activity is inhibited by S-nitrosylation of the cytosolic subunit p47phox, the organizer of the complex, reducing ROS production. Such NO-mediated suppression of NOX is compromised in atherosclerosis, primarily due to the reduced bioavailability of vascular NO (217, 218).
Xanthine Oxidase (XO).
Xanthine oxidase (XO) is an enzyme that catalyzes the oxidation of hypoxanthine (deaminated adenine) to xanthine and then xanthine to uric acid for purine catabolism. These reactions also generate superoxide and hydrogen peroxide as byproducts (219). In response to a stress, such as a high cholesterol level and metabolic changes, XO is released into the extracellular space or circulation and binds to glycosaminoglycans on the target cell surface, increasing the extracellular ROS levels (219, 220). XO-mediated ROS production is inhibited by its direct interaction with NOS1, suggesting the possible involvement of S-nitrosylation (221). In leptin-deficient obesity syndrome, NOS1 expression is reduced in the cardiac muscle, which overactivates XO to exacerbate oxidative stress, contributing to myocardial dysfunction (222).
Extracellular Superoxide Dismutase (Ec-SOD).
SOD catalyzes the dismutation of superoxide to peroxide and, in conjunction with catalase which converts peroxide to water, plays a major role in lowering ROS levels. Ec-SOD is a secretory SOD found in most tissues. Once secreted, it is localized in the glycocalyx (glycol-proteins and lipids covering the plasma membrane) of most types of cells, serving as a major extracellular antioxidant enzyme (199). Ec-SOD also binds to type I collagen in the extracellular matrix. Collagen is composed of 16 % prolines with which ROS could react to cause peptide bond cleavage. Ec-SOD binding confers protection to collagen against oxidative fragmentation (223). Ec-SOD also plays essential roles in determining the bioavailability of NO by preventing NO from reacting with superoxide to form peroxynitrite (224). However, Ec-SOD activity itself is influenced by neither NO nor S-nitrosylation (225).
Other Secretory Proteins Regulated by S-nitrosylation for Redox Control
Antioxidant Enzymes.
There are three major antioxidant enzymes, other than Ec-SOD, secreted into the extracellular environment to exert redox control. These enzymes are catalase (CAT), glutathione peroxidase (GPx) and thioredoxin (Trx). CAT catalyzes the decomposition of hydrogen peroxide to water and oxygen. GPx reduces hydrogen peroxide to water, while oxidizing glutathione to glutathione disulfide. Trx catalyzes the reduction of oxidized cysteines and removal of hydrogen peroxide. All the enzymes are targeted for S-nitrosylation, which inhibits their enzymatic activities to degrade hydrogen peroxide, possibly as a negative-feedback mechanism (200-202). In fact, the physiological level of hydrogen peroxide is necessary for certain types of redox signaling, whereas its excessive accumulation could confer oxidative stress (226). For example, the physiological level of hydrogen peroxide inactivates redox-regulated proteases, cathepsin and calcineurin, that degrade the extracellular matrix (ECM)(227, 228). On the other hand, the excessive levels (>100 nM) of hydrogen peroxide could oxidatively damage macromolecules, while sustained (>60 hours) production of hydrogen peroxide could activate matrix metalloproteinase (MMP) that degrades the ECM (226, 229).
Protein Disulfide Isomerase (PDI).
PDI is an enzyme that catalyzes the formation of disulfide bonds of proteins. PDI is secreted by platelets and endothelial cells during vascular injury to trigger thrombus formation. PDI is also upregulated in response to endoplasmic reticulum stress and disease conditions, such as cancer, serving as a chaperon to correct protein misfolding. Furthermore, PDI associates with NOX to promote the stability of the enzyme complex and the production of the extracellular ROS (230). PDI is targeted for S-nitrosylation to inhibit its enzymatic activity. S-nitrosylated PDI also serves as a transnitrosylase to propagate the S-nitrosyl signal. The consequence of S-nitrosylation of PDI depends on the context. In the healthy vasculature, where the optimal NO level is maintained, PDI is S-nitrosylated to suppress thrombus formation for the maintenance of vascular quiescence (231). On the other hand, in neurodegenerative diseases, where high levels of NO are produced, PDI is hyper-S-nitrosylated, which causes the over-accumulation of misfolded proteins and contributes to the disease pathogenesis (232).
DYSREGUALTED S-NITROSYLATION IN DISEASE PATHOGENESIS
S-nitrosylation is a key mechanism through which nitric oxide signal is propagated not only within the cell and tissue, but also in the microenvironment. Under normal physiological conditions, S-nitrosylation of proteins is highly regulated to maintain redox homeostasis. However when encountered with extreme hostile conditions, such as exposure to pathogens, radiation and carcinogens, this regulation could be interrupted, leading to the occurrence of diseases, most notably, cancer (18). Recent studies have indicated that S-nitrosylation is a major regulator of tumor microenvironment (TME), the surroundings of cancer cells that critically control the disease pathogenesis (130, 233, 234). Some of these proteins are listed in Table 1.
Tumor Microenvironment (TME) and Oxidative Stress
Tumor microenvironment (TME) is a dynamic network of cellular and acellular components that surround tumor cells (parenchyma) to form their niche. Cellular components of TME include stromal cells, such as fibroblasts, immune cells and adipocytes. Acellular components of TME include the extracellular matrix (ECM), growth factors, enzymes, cytokines and chemokines (235). Tumor cells and TME dynamically and reciprocally interact with each other to achieve their unique characteristics and to drive malignant progression (235-237). TME could be exacerbated by a multitude of intrinsic and extrinsic factors such as oxidative stress (236, 238, 239). ROS levels are elevated in tumor cells owing to various causes, including upregulation of ROS- producing enzymes or downregulation antioxidant enzymes (see above), increased basal metabolic activity, mitochondrial dysfunction, and peroxisome activity (239). The increased ROS levels not only cause damage in macromolecules such as DNA strand break, DNA-protein crosslinks, protein degradation (240), but also stimulate further production of ROS in TME. These mechanisms underlie the tumor supportive nature of TME and facilitate tumor progression (236, 238). In response to the increased level of ROS, cellular components of TME, especially, cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs), not only secrete various proteins that modulate TME, but also further produce ROS (234, 236, 238, 239, 241). Many of these proteins are targeted for S- nitrosylation, aiming to regulate the redox status of TME (238, 242). Among these, the major TME regulators are matrix metalloproteinases (MMPs) and tissue transglutaminase (TG2).
Matrix Metalloproteinases (MMPs).
MMPs are a family of endopeptidases, containing a Zn2+ in their catalytic site (128, 129, 133). They play key roles in ECM remodeling, wound healing, morphogenesis and host defense mechanism through their ability to cleave ECM and non-ECM substrates (128, 129, 243). In particular, overactivation of MMP-2, -3, -9 and -13 is implicated in carcinogenesis (129). Upon ROS production by CAFs and TAMs, MMPs are proteolytically activated (236, 239). MMPs then cleave ECM proteins and degrade cell surface molecules, such as E-cadherin and tissue transglutaminase. This causes ECM breakdown and dissociation of epithelial cells, promoting the mobility of cells in TME. MMPs also activate chemokines, cytokines (e.g. CXCL5, CXCL8) and growth factors (e.g. TGFβ) and degrade immune receptors (e.g. IL2 receptor), which altogether helps establish a pro-tumorigenic TME (243, 244). Especially, MMP9 promotes tumor growth, migration, as well as tumor-associated inflammation. MMP-9 cleaves and releases gelatin and type IV collagen, which compromises the ECM integrity and allows cancer stem cells to invade the ECM by binding to released ECM proteins (245). In contrast, MMP-9 also inhibits angiogenesis by releasing anti-angiogenic fragments, angiostatin and endostatin, attesting to the complexity of MMP9 functions (243, 246).
Cysteine residues in the conserved pro domain of MMP-9 interact with the Zn2+ ion, keeping the enzyme in an inactive form. These cysteine residues are targeted for S-nitrosylation. However, the consequences appear to be dependent on NO concentration. S-nitrosylation of these cysteines by low concentrations of NO exposes the Zn2+ ion and activates MMP-9 to promote tumorigenesis, a phenomenon termed the cysteine switch (128, 129, 233, 243, 247). In contrast, S-nitrosylation of these cysteines by higher concentrations of NO (for example, during inflammation) is proposed to contribute to the release of the Zn2+ ion from the active site and inactivation of MMP-9 (18, 128, 244, 247). Although the detailed underlying mechanism has yet to be clarified, such S-nitrosylation-mediated inhibition of MMP-9 bears a therapeutic potential for cancer.
Tissue Transglutaminase (TG2).
Transglutaminases (TGs) are a family of multi–functional enzymes that catalyze transamidation of proteins in a Ca2+ dependent manner (122, 248). Among the nine members of TG family, tissue transglutaminase (TG2) is a versatile enzyme expressed in many different types of cells in TME, including smooth muscle cells, endothelial cells fibroblasts and TAMs. TG2 influences tumor growth, metastasis, apoptosis and chemo-resistance (122, 123, 249). TG2’s activities are based on its subcellular localization. Membrane-bound TG2 hydrolyzes GTP and mediates signal transduction for cell cycle progression. In contrast, secreted or cytosolic TG2 crosslinks proteins either by forming stable N-ε-(γ- glutamyl) lysine isopeptide bonds or by incorporating polyamines (122, 123, 249-251). TG2 crosslinks different ECM proteins, such as collagen, fibronectin and osteonectin, to stabilize the ECM architecture (248, 250, 251). TG2 also induces TGFβ activation, leading to the deposition of ECM proteins (123, 250). Furthermore, TG2 elicits anti-apoptotic functions through Caspase 3 inhibition, NF-kB activation and crosslinking (inhibition) of retinoblastoma protein (pRb) during nuclear translocation (248, 250, 251).
TG2, harboring 18 cysteine residues, undergoes poly S-nitrosylation in a Ca2+ dependent manner (249). S–nitrosylation of Cys277 is proposed to inhibit its transamidation activity (123, 251). Since TG2 is implicated in tumorigenesis, this S-nitrosylation-mediated inhibition of TG2 could be utilized in anti-cancer therapy.
Conclusions
Cumulative evidence suggests that S-nitrosylation plays a large part in NO-mediated biological activities (11). On one hand, basal S-nitrosylation takes place at the physiological, steady-state level of NO and contributes to the maintenance of homeostasis (10, 15, 31, 203, 252-255). On the other hand, S-nitrosylation could be induced as a sensor of an oxidizing condition, such as hyperoxia or increased ROS levels. The nitrosyl group could travel to different subcellular locales through a cascade of transnitrosylation reactions (58). S-nitrosylation is an emerging paradigm of redox signaling that could simultaneously control a large number of proteins involved in divergent pathways. S-nitrosylation helps stop further ROS production, protect cellular proteins against oxidative damage and propagate redox signaling. The homeostatic level of S-nitrosylation is controlled by the balance between S-nitrosylation and denitrosylation which are spatially and temporally regulated. However, S-nitrosylation levels are dysregulated in many different types of diseases, such as neurodegenerative diseases and cancer, contributing to the disease pathogenesis. Whether restoration of the homeostatic level of S-nitrosylation might serve as an effective therapeutic intervention for certain diseases would warrant active investigation.
Author contributions: Conceptualization, S.F.; Original Draft Preparation, S.F., V.F., Y.W., X.Z., and V.S.; Writing, Review & Editing, S.F., V.F, Y.W, J.L. and V,S.; Figures: S.F., Table: V.F. and S.F.; and Funding Acquisition, S.F.
Conflicts of Interest statement: The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Furchgott RF, Cherry PD, Zawardzki JV, Jothianandan D. Endothelial cells as mediators of vasodilation of arteries. J Cardiovasc Pharmacol. 1984;6:S336–S43.
- Cudeiro J, Rivadulla C, Rodríguez R, Martínez-Conde S, Martínez L, Grieve KL, Acuña C. Further observations on the role of nitric oxide in the feline lateral geniculate nucleus. Eur J Neurosci. 1996;8:144-52.
- Ho GP, Selvakumar B, Mukai J, Hester LD, Y. W, Gogos JA, Snyder SH. S-nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron. 2011;71(1):131-41.
- Martínez-Ruiz A, Villanueva L, González de Orduña C, López-Ferrer D, Higueras MA, Tarín C, Rodríguez-Crespo I, Vázquez J, Lamas S. S-nitrosylation of Hsp90 promotes the inhibition of its ATPase and endothelial nitric oxide synthase regulatory activities. Proc Natl Acad Sci U S A. 2005;102(24):8525-30.
- Nakamura T, Lipton SA. Protein S-Nitrosylation as a Therapeutic Target for Neurodegenerative Diseases. Trends Pharmacol Sci. 2016;37(1):73-84.
- Thomas DD, Liu X, Kantrow SP, Lancaster JRJ. The biological lifetime of nitric oxide: implications for the perivascular dynamics of NO and O2. Proc Natl Acad Sci U S A. 2001;98(1):355-60.
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357(Pt3):593-615.
- Wink DA, Miranda KM, Espey MG, Pluta RM, Hewett SJ, Colton C, Vitek M, Feelisch M, Grisham MB. Mechanisms of the antioxidant effects of nitric oxide. Antioxidants & redox signaling. 2001;3(2):203-13.
- Luiking YC, Engelen MP, Deutz NE. Regulation of nitric oxide production in health and disease. Curr Opin Clin Nutr Metab Care. 2010;13(1):97-104.
- Francis SH, Busch JL, Corbin JD, Sibley D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev. 2010;62(3):525-63.
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6(2):150--66.
- Hess DT, Stamler JS. Regulation by S-nitrosylation of protein post-translational modification. J Bioil Chem. 2012;287(7):4411-8.
- Selvakumar B, Jenkins MA, Hussain NK, Huganir RL, Traynelis SF, Snyder SH. S-nitrosylation of AMPA receptor GluA1 regulates phosphorylation, single-channel conductance, and endocytosis. Proc Nat Acad Sci U S A. 2013;110(3):1077-82.
- Martínez-Ruiz A, Lamas S. S-nitrosylation: a potential new paradigm in signal transduction. Cardiovasc Res. 2004;62(1).
- Sun J, Steenbergen C, Murphy E. S-nitrosylation: NO-related redox signaling to protect against oxidative stress. Antioxid Redox Signal. 2006;8(9-10):1693-705.
- Jerca L, Jerca O, Mancas G, Constantinescu I, Lupuşoru R. MECHANISM OF ACTION AND BIOCHEMICAL EFFECTS OF NITRIC OXIDE J Prev Med. 2002;10(2):35-45.
- Kovacs I, Lindermayr C. Nitric oxide-based protein modification: formation and site-specificity of protein S-nitrosylation. Front Plant Sci. 2013;4:137.
- Wang Z. Protein S-nitrosylation and cancer. Cancer Lett. 2012;320(2):123-9.
- Plenchette S, Romagny S, Laurens V, Bettaieb A. S-Nitrosylation in TNF superfamily signaling pathway: Implication in cancer. Redox Biol. 2015;6:507-15.
- Antosova M, Plevkova J, Strapkova A, Buday T. Nitric oxide—Important messenger in human body. OJMIP. 2012;2:98-106.
- Peunova N, Scheinker V, Ravi K, Enikolopov G. Nitric oxide coordinates cell proliferation and cell movements during early development of Xenopus. Cell Cycle. 2007;6:3132-44.
- Young SL, Evans K, Eu JP. Nitric oxide modulates branching morphogenesis in fetal rat lung explants. Am J Physiol Lung Cell Mol Physiol. 2002;282(3):L379-85.
- Cole AG, Mashkournia A, Parries SC, Goldberg JI. Regulation of early embryonic behavior by nitric oxide in the pond snail Helisoma trivolvis. J Exp Biol. 2002;205(Pt 20):3143-52.
- Slezinger MS, Kuzin BA. Nitric oxide synthase mediates regulation of cell polarity and movement during Drosophila melanogaster morphogenesis. Ontogenez. 2009;40(1):40-7.
- Bradley S, Tossell K, Lockley R, McDearmid JR. Nitric oxide synthase regulates morphogenesis of zebrafish spinal cord motoneurons. J Neurosci. 2010;30(50):16816-31.
- Lee NP, Cheng CY. Nitric oxide/nitric oxide synthase, spermatogenesis, and tight junction dynamics. Biol Reprod. 2004;70(2):267-76.
- Nunoshiba T, DeRojas-Walker T, Wishnok JS, Tannenbaum SR, Demple B. Activation by nitric oxide of an oxidative-stress response that defends Escherichia coli against macrophages. Proc Natl Acad Sci U S A. 1993;90:9993–7.
- Kim YM, Bergonia H, Lancaster JRJ. Nitrogen oxide-induced autoprotection in isolated rat hepatocytes. FEBS Lett. 1995;374:228–23.
- Clancy RM, Leszczynska-Piziak J, Abramson SB. Nitric oxide, an endothelial cell relaxation factor, inhibits neutrophil superoxide anion production via a direct action on the NADPH oxidase. J Clin Invest. 1992;90:1116–21.
- Bice JS, Jones BR, Chamberlain GR, Baxter GF. Nitric oxide treatments as adjuncts to reperfusion in acute myocardial infarction: a systematic review of experimental and clinical studies. Basic Res Cardiol 2016;111(2):23.
- Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Hussain P, Vecoli C, Paolocci N, Ambs S, Colton CA, Harris CC, Roberts DD, Wink DA. The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med. 2008;45(1):18-31.
- Loughran PA, Stolz DB, Barrick SR, Wheeler DS, Friedman PA, Rachubinski RA, Watkins SC, Billiar TR. PEX7 and EBP50 target iNOS to the peroxisome in hepatocytes. Nitric Oxide. 2013;31:9-19.
- Prolo C, Alvarez MN, Radi R. Peroxynitrite, a potent macrophage-derived oxidizing cytotoxin to combat invading pathogens. Biofactors. 2014;40(2):215-25.
- Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Hear J. 2012;33(7):829-37.
- Lacza Z, Puskar M, Figueroa JP, Zhang J, Rajapakse N, Busija DW. Mitochondrial nitric oxide synthase is constitutively active and is functionally upregulated in hypoxia. Free Radic Biol Med. 2001;31(12):1609-15.
- Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26(4):190-5.
- Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310(5746):314-7.
- Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv Z, Gu Z, Zhang J, Wang J, Zen K, Xiang Y, Wang D, Zhang CY. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. 2010;20(6):676-87.
- Victor VM, Nun˜ez C, D’Oco´n P, Taylor CT, Esplugues JV, Moncada S. Regulation of oxygen distribution in tissues by endothelial nitric oxide. Circ Res. 2009;104:1178-83.
- Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26(11):2439-44.
- Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279(36):37918-27.
- Andrew PJ, Mayer B. Enzymatic function of nitric oxide synthases. Cardiovasc Res 1999;43(5):521-31.
- Rubio-Guerra AF, Vargas-Robles H, Ramos-Brizuela LM, Escalante-Acosta BA. Is tetrahydrobiopterin a therapeutic option in diabetic hypertensive patients? Integr Blood Press Control 2010;3:125-32.
- Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. The Journal of Clinical Investigation. 2003;111(8):1201-9. doi: 10.1172/JCI14172.
- Hoang HH, Padgham SV, Meininger CJ. L-arginine, tetrahydrobiopterin, nitric oxide and diabetes. Curr Opin Clin Nutr Metab Care. 2013;16(1):76-82.
- Gámez-Méndez AM, Vargas-Robles H, Arellano-Mendoza M, Cruz-Laguna E, Rios A, Escalante B. Early stage of obesity potentiates nitric oxide reduction during the development of renal failure. J Nephrol. 2014;27(3):281-7.
- Rabender CS, Alam A, Sundaresan G, Cardnell RJ, Yakovlev VA, Mukhopadhyay ND, Graves P, Zweit J, Mikkelsen RB. The Role of Nitric Oxide Synthase Uncoupling in Tumor Progression. Mol Cancer Res. 2015;13(6):1034-43.
- Veron D, Aggarwal PK, Velazquez H, Kashgarian M, Moeckel G, Tufro A. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J Am Soc Nephrol. 2014;25(8):1814-24.
- Seo BR, Bhardwaj P, Choi S, Gonzalez J, Andresen Eguiluz RC, Wang K, Mohanan S, Morris PG, Du B, Zhou XK, Vahdat LT, Verma A, Elemento O, Hudis CA, Williams RM, Gourdon D, Dannenberg AJ, Fischbach C. Obesity-dependent changes in interstitial ECM mechanics promote breast tumorigenesis. Sci Transl Med. 2015;7(301):301ra130.
- Edgar KS, Matesanz N, Gardiner TA, Katusic ZS, McDonald DM. Hyperoxia depletes (6R)-5,6,7,8-tetrahydrobiopterin levels in the neonatal retina: implications for nitric oxide synthase function in retinopathy. Am J Pathol. 2015;185(6):1769-82.
- Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. The Journal of biological chemistry. 2003;278(25):22546-54.
- Werner-Felmayer G, Golderer G, Werner ER. Tetrahydrobiopterin biosynthesis, utilization and pharmacological effects. Curr Drug Metab. 2002;3(2):159-73.
- Kim HL, Park YS. Maintenance of cellular tetrahydrobiopterin homeostasis. BMB Rep. 2010;43(9):584-92.
- Latremoliere A, Costigan M. GCH1, BH4 and pain. Curr Pharm Biotechnol. 2011;12(10):1728-41.
- Shintaku H. Disorders of tetrahydrobiopterin metabolism and their treatment. Curr Drug Metab. 2002;3(2):123-31.
- Martínez-Ruiz A, Araújo IM, Izquierdo-Álvarez A, Hernansanz-Agustín P, Lamas S, Serrador J. Specificity in S-nitrosylation: a short-range mechanism for NO signaling? Antioxidants & redox signaling. 2013;19(11):1220-35.
- Figueroa XF, Lillo MA, Gaete PS, Riquelme MA, Sáez JC. Diffusion of nitric oxide across cell membranes of the vascular wall requires specific connexin-based channel. Neuropharmacology. 2013;75:471-8.
- Nakamura T, Lipton SA. Emerging role of protein-protein transnitrosylation in cell signaling pathways. Antioxid Redox Signal. 2013;18(3):239-49.
- Chang AH, Sancheti H, Garcia J, Kaplowitz N, Cadenas E, Han D. Respiratory substrates regulate S-nitrosylation of mitochondrial proteins through a thiol-dependent pathway. Chem Res Toxicol. 2014;27(5):794-804.
- Piantadosi CA. Regulation of Mitochondrial Processes by Protein S-Nitrosylation. Biochimica et Biophysica Acta. 2012;1820(6):712-21. doi: 10.1016/j.bbagen.2011.03.008. PubMed PMID: PMC3162082.
- Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci U S A. 1998;95:7631–6.
- Penna C, Sorge M, Femminò S, Pagliaro P, Brancaccio M. Redox Aspects of Chaperones in Cardiac Function. Front Physiol. 2018;9:216.
- Rossi-George A, Gow A. Nitric Oxide Biochemistry: Pathophysiology of Nitric Oxide-Mediated Protein Modifications. SC Veasey (ed), Oxidative Neural Injury, Contemporary Clinical Neuroscience, Humana Press. 2009:29-44.
- Oae S, Shinhama K. Organic thionitrites and related substances. Org Prep Proc Int. 1983;16:165-98.
- Stamler J, S,, Jaraki O, Osborne J, Simon D, I,, Keaney j, Vita J, Singel D, Valeri CR, Loscalzo J. Nitric oxide circulates in mammalian plasma primarily as an S-nitroso adduct of serum albumin. Proc Natl Acad Sci U S A. 1992;88:7674–7.
- Anand P, Stamler J. Enzymatic mechanisms regulating protein S-nitrosylation: implications in health and disease. J Mol Med (Berl). 2012;90(3):233-44.
- Keilin D, Hartree EF. Reaction of nitric oxide with haemoglobin and methaemoglobin. Nature. 1937;139:548.
- Murad F. Regulation of cytosolic guanylyl cyclase by nitric oxide: the NO- cyclic GMP signal transduction system. Adv Pharmacol. 1994;26:19-33.
- Sarti P, Forte E, Mastronicola D, Giuffrè A, Arese M. Cytochrome c oxidase and nitric oxide in action: molecular mechanisms and pathophysiological implications. Biochim Biophys Acta. 2012;1817(4):610-9.
- Fago A, Crumbliss AL, Hendrich MP, Pearce LL, Peterson J, Henkens R, Bonaventura C. Oxygen binding to partially nitrosylated hemoglobin. Biochim Biophys Acta. 2013;1834(9):1894-900.
- Jung T, Höhn A, Grune T. The proteasome and the degradation of oxidized proteins: Part II - protein oxidation and proteasomal degradation. Redox Biol. 2014;2:99-104.
- Freeman BA, Baker PR, Schopfer FJ, Woodcock SR, Napolitano A, d'Ischia M. Nitro-fatty acid formation and signaling. J Biol Chem. 2008;283(23):15515-9.
- Lancaster JRJ. Nitric oxide: a brief overview of chemical and physical properties relevant to therapeutic applications. Future Sci OA. 2015;1(1):FSO59.
- Ravi K, Brennan LA, Levic S, Ross PA, Black SM. S-nitrosylation of endothelial nitric oxide synthase is associated with monomerization and decreased enzyme activity. Proc Natl Acad Sci U S A. 2004;101(8):2619-24.
- Thapliyal A, Verma R, Kumar N. Small G Proteins Dexras1 and RHES and Their Role in Pathophysiological Processes. Int J Cell Biol. 2014;2014:308535.
- Trepel J, Mollapour M, Giaccone G, Neckers L. Targeting the dynamic HSP90 complex in cancer. Nat Rev Cancer 2010;10(8):537-49.
- Hsu K, Champaiboon C, Guenther BD, Sorenson BS, Khammanivong A, Ross KF, Geczy CL, Herzberg MC. ANTI-INFECTIVE PROTECTIVE PROPERTIES OF S100 CALGRANULINS. Antiinflamm Antiallergy Agents Med Chem. 2009;8(4):290-305.
- Jia J, Arif A, Terenzi F, Willard B, Plow EF, Hazen SL, Fox PL. Target-selective protein S-nitrosylation by sequence motif recognition. Cell. 2014;159(3):623-34.
- Lim SY, Raftery M, Cai H, Hsu K, Yan WX, Hseih HL, Watts RN, Richardson D, Thomas S, Perry M, Geczy CL. S-nitrosylated S100A8: novel anti-inflammatory properties. J Immunol. 2008;181(8):5627-36.
- Mendez MG, Kojima S, Goldman RD. JVimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB 2010;26(6):1838-51.
- Fehon RG, McClatchey A, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11(4):276-87.
- Pérez-Mato I, Castro C, Ruiz FA, Corrales FJ, Mato JM. Methionine adenosyltransferase S-nitrosylation is regulated by the basic and acidic amino acids surrounding the target thiol. J Biol Chem 1999;274(24):17075-9.
- Doulias PT, Greene JL, Greco TM, Tenopoulou M, Seeholzer SH, Dunbrack RL, Ischiropoulos H. Structural profiling of endogenous S-nitrosocysteine residues reveals unique features that accommodate diverse mechanisms for protein S-nitrosylation. Proc Natl Acad Sci U S A. 2010;107(39):16958-63.
- Möller MN, Li Q, Vitturi DA, Robinson JM, Lancaster JRJ, Denicola A. Membrane "lens" effect: focusing the formation of reactive nitrogen oxides from the *NO/O2 reaction. Chem Res Toxicol. 2007;20(4):709-14.
- Broniowska KA, Hogg N. The chemical biology of S-nitrosothiols. Antioxid Redox Signal. 2012;17(7):969-80.
- Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proc Natl Acad Sci U S A. 2002;99(17):10958-63.
- Cheng S, Shi T, Wang XL, Liang J, Wu H, Xie L, Li Y, Zhao YL. Features of S-nitrosylation based on statistical analysis and molecular dynamics simulation: cysteine acidity, surrounding basicity, steric hindrance and local flexibility. Mol Biosyst. 2014;10(10):2597-606.
- Beltrán B, Orsi A, Clementi E, Moncada S. Oxidative stress and S-nitrosylation of proteins in cells. Br J Pharmacol. 2000;129(5):953-60.
- Butzer U, Weidenbach H, Gansauge S, Gansauge F, Beger HG, Nussler AK. Increased oxidative stress in the RAW 264.7 macrophage cell line is partially mediated via the S-nitrosothiol-induced inhibition of glutathione reductase. FEBS Lett. 1999;445(2-3):274-8.
- Whiteman M, Chua YL, Zhang D, Duan W, Liou YC, Armstrong JS. Nitric oxide protects against mitochondrial permeabilization induced by glutathione depletion: role of S-nitrosylation? Biochem Biophys Res Commun. 2006;339(1):255-62.
- Stomberski CT, Hess DT, Stamler JS. Protein S-Nitrosylation: Determinants of Specificity and Enzymatic Regulation of S-Nitrosothiol-Based Signaling. Antioxidants & redox signaling. 2019;30(10):1331-51.
- Kovacs I, Holzmeister C, Wirtz M, Geerlof A, Fröhlich T, Römling G, Kuruthukulangarakoola GT, Linster E, Hell R, Arnold GJ, Durner J, Lindermayr C. ROS-Mediated Inhibition of S-nitrosoglutathione Reductase Contributes to the Activation of Anti-oxidative Mechanisms. Front Plant Sci. 2016;7:1669.
- Zhang Y, Wu K, Su W, Zhang DF, Wang P, Qiao X, Yao Q, Yuan Z, Yao YG, Liu G, Zhang C, Liu L, Chen C. Increased GSNOR Expression during Aging Impairs Cognitive Function and Decreases S-Nitrosation of CaMKIIα. J Neurosci. 2017;37(40):9741-58.
- Caplin B, Leiper J. Endogenous nitric oxide synthase inhibitors in the biology of disease: markers, mediators, and regulators? Arterioscler Thromb Vasc Biol. 2012;32(6):1343-53.
- Fan JS, Zhang Q, Li M, Tochio H, Yamazaki T, Shimizu M, Zhang M. Protein inhibitor of neuronal nitric-oxide synthase, PIN, binds to a 17-amino acid residue fragment of the enzyme. The Journal of biological chemistry. 1998;273(50):33472-81.
- Broniowska KA, Keszler A, Basu S, Kim-Shapiro DB, Hogg N. Cytochrome c-mediated formation of S-nitrosothiol in cells. Biochem J. 2012;442(1):191-7.
- Dahm CC, Moore K, Murphy MP. Persistent S-nitrosation of complex I and other mitochondrial membrane proteins by S-nitrosothiols but not nitric oxide or peroxynitrite: implications for the interaction of nitric oxide with mitochondria. J Biiol Chem. 2006;281:0056–10065.
- Giri S, Rattan R, Deshpande M, Maguire JL, Johnson Z, Graham RP, Shridhar V. Preclinical therapeutic potential of a nitrosylating agent in the treatment of ovarian cancer. PLos One. 2014;9(6):e97897.
- Furuhashi S, Sugita H, Takamori H, Horino K, Nakahara O, Okabe H, Miyake K, Tanaka H, Beppu T, Baba H. NO donor and MEK inhibitor synergistically inhibit proliferation and invasion of cancer cells. Int J Oncol. 2012;40(3):807-15.
- Olson N, Kasahara D, Hristova M, Bernstein R, Janssen-Heininger Y, van der Vliet A. Modulation of NF-κB and hypoxia-inducible factor--1 by S-nitrosoglutathione does not alter allergic airway inflammation in mice. Am J Respir Cell Mol Biol. 2011;44(6):813-23.
- Kaliyaperumal K, Sharma AK, McDonald DG, Dhindsa JS, Yount C, Singh AK, Won JS, Singh I. S-Nitrosoglutathione-mediated STAT3 regulation in efficacy of radiotherapy and cisplatin therapy in head and neck squamous cell carcinoma. Redox Biol. 2015;6(41-50).
- Marozkina NV, Gaston B. S-Nitrosylation signaling regulates cellular protein interactions. Biochim Biophys Acta 2012;1820(6):722-9.
- Karin M. NF-κB as a Critical Link Between Inflammation and Cancer. Cold Spring Harb Perspect Bioil. 2009;1(5):a000141.
- Tristan C, Shahani N, Sedlak TW, Sawa A. The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal. 2011;23(2):317-23.
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7(7):665-74.
- Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12(11):1094-100.
- Reed JC, Ely KR. Degrading liaisons: Siah structure revealed. Nat Struct Biol. 2002;9(1):8-10.
- Zhai D, Chin K, Wang M, Liu F. Disruption of the nuclear p53-GAPDH complex protects against ischemia-induced neuronal damage. Mol Brain. 2014;7:20.
- Thangima Zannat M, Bhattacharjee RB, Bag J. In the absence of cellular poly (A) binding protein, the glycolytic enzyme GAPDH translocated to the cell nucleus and activated the GAPDH mediated apoptotic pathway by enhancing acetylation and serine 46 phosphorylation of p53. Biochem Biophys Res Commun. 2011;409:171-6.
- Kohr MJ, Murphy E, Steenbergen C. Glyceraldehyde-3-phosphate dehydrogenase acts as a mitochondrial trans-S-nitrosylase in the heart. PLoS One. 2014;9(10):e111448.
- Nakajima H, Itakura M, Kubo T, Kaneshige A, Harada N, Izawa T, Azuma Y-T, Kuwamura M, Yamaji R, Takeuchi T. Glyceraldehyde-3-phosphate Dehydrogenase (GAPDH) Aggregation Causes Mitochondrial Dysfunction during Oxidative Stress-induced Cell Death. Journal of Biological Chemistry. 2017;292(11):4727-42. doi: 10.1074/jbc.M116.759084.
- Wen YY, Yang ZQ, Song M, Li BL, Yao XH, Chen XL, Zhao J, Lu YY, Zhu JJ, Wang EH. The expression of SIAH1 is downregulated and associated with Bim and apoptosis in human breast cancer tissues and cells. Mol Carcinog. 2010;49(5):440-9.
- Zhang JY, Zhang F, Hong CQ, Giuliano AE, Cui XJ, Zhou GJ, Zhang GJ, Cui YK. Critical protein GAPDH and its regulatory mechanisms in cancer cells. Cancer Biol Med. 2015;12(1):10-22.
- Fiucci G, Beaucourt S, Duflaut D, Lespagnol A, Stumptner-Cuvelette P, Géant A, Buchwalter G, Tuynder M, Susini L, Lassalle JM, Wasylyk C, Wasylyk B, Oren M, Amson R, Telerman A. Siah-1b is a direct transcriptional target of p53: identification of the functional p53 responsive element in the siah-1b promote. Proc Natl Acad Sci U S A. 2004;101(10):3510-5.
- Tardif MR, Chapeton-Montes JA, Posvandzic A, Pagé N, Gilbert C, Tessier PA. Secretion of S100A8, S100A9, and S100A12 by Neutrophils Involves Reactive Oxygen Species and Potassium Efflux. J Immunol Res. 2015;2015:296149.
- Kerkhoff C, Nacken W, Benedyk M, Dagher MC, Sopalla C, Doussiere J. The arachidonic acid-binding protein S100A8/A9 promotes NADPH oxidase activation by interaction with p67phox and Rac-2. FASAB J. 2005;19(3):467-9.
- Biri-Kovács B, Kiss B, Vadászi H, Gógl G, Pálfy G, Török G, Homolya L, Bodor A, Nyitray L. Ezrin interacts with S100A4 via both its N- and C-terminal domains. PLoS One. 2017;12(5):v.
- Baritaki S, Huerta-Yepez S, Sahakyan A, Karagiannides I, Bakirtzi K, Jazirehi A, Bonavida B. Mechanisms of nitric oxide-mediated inhibition of EMT in cancer: inhibition of the metastasis-inducer Snail and induction of the metastasis-suppressor RKIP. Cell Cycle. 2010;9(24):4931-40.
- Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154(6):1111-6. Epub 2001/09/10. doi: 10.1083/jcb.200104008. PubMed PMID: 11551979.
- Plenchette S, Romagny S, Laurens V, Bettaieb A. S-Nitrosylation in TNF superfamily signaling pathway: Implication in cancer. Redox Biol. 2015;6:507-15. Epub 2015/10/09. doi: 10.1016/j.redox.2015.08.019. PubMed PMID: 26448396; PMCID: PMC4600855.
- Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, Takahashi M, Cheah JH, Tankou SK, Hester LD, Ferris CD, Hayward SD, Snyder SH, Sawa A. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nature Cell Biology. 2005;7(7):665-74. doi: 10.1038/ncb1268.
- Santhanam L, Tuday EC, Webb AK, Dowzicky P, Kim JH, Oh YJ, Sikka G, Kuo M, Halushka MK, Macgregor AM, Dunn J, Gutbrod S, Yin D, Shoukas A, Nyhan D, Flavahan NA, Belkin AM, Berkowitz DE. Decreased S-nitrosylation of tissue transglutaminase contributes to age-related increases in vascular stiffness. Circ Res. 2010;107(1):117-25. Epub 2010/05/22. doi: 10.1161/CIRCRESAHA.109.215228. PubMed PMID: 20489165.
- Telci D, Collighan RJ, Basaga H, Griffin M. Increased TG2 expression can result in induction of transforming growth factor beta1, causing increased synthesis and deposition of matrix proteins, which can be regulated by nitric oxide. J Biol Chem. 2009;284(43):29547-58. Epub 2009/08/07. doi: 10.1074/jbc.M109.041806. PubMed PMID: 19657147; PMCID: PMC2785588.
- Tang C-H, Wei W, Liu L. Regulation of DNA repair by S-nitrosylation. Biochimica et biophysica acta. 2012;1820(6):730-5. Epub 2011/05/05. doi: 10.1016/j.bbagen.2011.04.014. PubMed PMID: 21571039.
- Benhar M. Emerging Roles of Protein S-Nitrosylation in Macrophages and Cancer Cells. Curr Med Chem. 2016;23(24):2602-17. Epub 2016/10/26. doi: 10.2174/0929867323666160627. PubMed PMID: 27356534.
- Nott A, Nitarska J, Veenvliet JV, Schacke S, Derijck AAHA, Sirko P, Muchardt C, Pasterkamp RJ, Smidt MP, Riccio A. S-nitrosylation of HDAC2 regulates the expression of the chromatin-remodeling factor Brm during radial neuron migration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(8):3113-8. Epub 2013/01/28. doi: 10.1073/pnas.1218126110. PubMed PMID: 23359715.
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-nitrosylation of histone deacetylase 2 induces chromatin remodelling in neurons. Nature. 2008;455:411. doi: 10.1038/nature07238
https://www.nature.com/articles/nature07238#supplementary-information.
- Gu Z, Kaul M, Yan B, Kridel SJ, Cui J, Strongin A, Smith JW, Liddington RC, Lipton SA. S-nitrosylation of matrix metalloproteinases: signaling pathway to neuronal cell death. Science. 2002;297(5584):1186-90. Epub 2002/08/17. doi: 10.1126/science.1073634. PubMed PMID: 12183632.
- O'Sullivan S, Medina C, Ledwidge M, Radomski MW, Gilmer JF. Nitric oxide-matrix metaloproteinase-9 interactions: biological and pharmacological significance--NO and MMP-9 interactions. Biochim Biophys Acta. 2014;1843(3):603-17. Epub 2013/12/18. doi: 10.1016/j.bbamcr.2013.12.006. PubMed PMID: 24333402.
- Bignon E, Allega MF, Lucchetta M, Tiberti M, Papaleo E. Computational Structural Biology of S-nitrosylation of Cancer Targets. Front Oncol. 2018;8:272. Epub 2018/08/30. doi: 10.3389/fonc.2018.00272. PubMed PMID: 30155439; PMCID: PMC6102371.
- Thibeault S, Rautureau Y, Oubaha M, Faubert D, Wilkes BC, Delisle C, Gratton J-P. S-Nitrosylation of β-Catenin by eNOS-Derived NO Promotes VEGF-Induced Endothelial Cell Permeability. Molecular Cell. 2010;39(3):468-76. doi: https://doi.org/10.1016/j.molcel.2010.07.013.
- Nakamura T, Prikhodko OA, Pirie E, Nagar S, Akhtar MW, Oh C-K, McKercher SR, Ambasudhan R, Okamoto S-i, Lipton SA. Aberrant protein S-nitrosylation contributes to the pathophysiology of neurodegenerative diseases. Neurobiol Dis. 2015;84:99-108. Epub 2015/03/18. doi: 10.1016/j.nbd.2015.03.017. PubMed PMID: 25796565.
- Martinez-Ruiz A, Lamas S. S-nitrosylation: a potential new paradigm in signal transduction. Cardiovasc Res. 2004;62(1):43-52. Epub 2004/03/17. doi: 10.1016/j.cardiores.2004.01.013. PubMed PMID: 15023551.
- Perez-Mato I, Castro C, Ruiz FA, Corrales FJ, Mato JM. Methionine adenosyltransferase S-nitrosylation is regulated by the basic and acidic amino acids surrounding the target thiol. Journal of Biological Chemistry. 1999;274(24):17075-9. doi: DOI 10.1074/jbc.274.24.17075. PubMed PMID: WOS:000080780400059.
- Casero RA, Murray Stewart T, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nature Reviews Cancer. 2018;18(11):681-95.
- Zhu L, Zhang C, Liu Q. PTEN S-nitrosylation by NOS1 inhibits autophagy in NPC cells. Cell Death & Disease. 2019;10(4):306. doi: 10.1038/s41419-019-1542-0.
- Marshall HE, Foster MW. S-nitrosylation of Ras in breast cancer. Breast Cancer Res. 2012;14(6):113-. doi: 10.1186/bcr3331. PubMed PMID: 23167903.
- Robb EL, Page MM, Stuart JA. Mitochondria, cellular stress resistance, somatic cell depletion and lifespan. Curr Aging Sci. 2012;2(1):12-27.
- Nakamura T, Lipton S. Cell death: protein misfolding and neurodegenerative diseases. Apoptosis. 2009;14:455–68.
- Ozawa K, Komatsubara AT, Nishimura Y, Sawada T, Kawafune H, Tsumoto H, Tsuji Y, Zhao J, Kyotani Y, Tanaka T, Takahashi R, Yoshizumi M. S-nitrosylation regulates mitochondrial quality control via activation of parkin. Sci Rep. 2013;3:2202.
- Mailloux RJ, Jin X, Willmore WG. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2013;2:129-39.
- Doulias PT, Tenopoulou M, Greene JL, Raju K, Ischiropoulos H. Nitric oxide regulates mitochondrial fatty acid metabolism through reversible protein S-nitrosylation. Sci Signal. 2013;6(256):rs1.
- Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. I Biol Chem. 2011;286(46):40184-92.
- Mannick JB, Schonhoff C, Papeta N, Ghafourifar P, Szibor M, Fang K, Gaston B. S-Nitrosylation of mitochondrial caspases. J Cell Biol. 2001;154(6):11111-6.
- Azad N, Vallyathan V, Wang L, Tantishaiyakul V, Stehlik C, Leonard SS, Rojanasakul Y. S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal degradation. A novel antiapoptotic mechanism that suppresses apoptosis. The Journal of biological chemistry. 2006;281(45):34124-34.
- Brown GC, Borutaite V. Inhibition of mitochondrial respiratory complex I by nitric oxide, peroxynitrite and S-nitrosothiols. Biochim Biophys Acta. 2004;1658(1-2):44-9.
- Hroudová J, Fišar Z. Control mechanisms in mitochondrial oxidative phosphorylation. Neural Regen Res. 2013;8(4):363-75.
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417(1):1-13.
- Zhang J, Jin B, Li L, Block ER, Patel JM. Nitric oxide-induced persistent inhibition and nitrosylation of active site cysteine residues of mitochondrial cytochrome-c oxidase in lung endothelial cells. Am J Physiol Cell Physiol. 2005;288(4):C840-9.
- Le Cras TD, McMurtry IF. Nitric oxide production in the hypoxic lung. Am J Physiol Lung Cell Mol Physiol. 2001;280(4):L575-82.
- Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302(5652):1975-8.
- Zorova LD, Popkov VA, Plotnikov EY, Silachev DN, Pevzner IB, Jankauskas SS, Babenko VA, Zorov SD, Balakireva AV, Juhaszova M, Sollott SJ, Zorov DB. Mitochondrial membrane potential. Anal Biochem. 2018;552:50-9.
- Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res. 2006;72(2):210-9.
- Basu S, Keszler A, Azarova NA, Nwanze N, Perlegas A, Shiva S, Broniowska KA, Hogg N, Kim-Shapiro DB. A novel role for cytochrome c: Efficient catalysis of S-nitrosothiol formation. Free Radic Biol Med. 2010;48(2):255-63.
- Schonhoff CM, Gaston B, Mannick JB. Nitrosylation of cytochrome c during apoptosis. J Biol Chem. 2003;278(20):18265-70.
- Liemburg-Apers DC, Willems PH, Koopman WJ, Grefte S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch Toxicol. 2015;89(8):1209-26.
- Speijer D, Manjeri GR, Szklarczyk R. How to deal with oxygen radicals stemming from mitochondrial fatty acid oxidation. Philos Trans R Soc Lond B Biol Sci 2014(369):1646.
- Quijano C, Trujillo M, Castro L, Trostchansky A. Interplay between oxidant species and energy metabolism. Redox Biol,. 2016;8:28-42.
- Quinlan CL, Perevoshchikova IV, Hey-Mogensen M, Orr AL, Brand MD. Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 2013;1:304-12.
- Chouchani ET, Hurd TR, Nadtochiy SM, Brookes PS, Fearnley IM, Lilley KS, Smith RA, Murphy MP. Identification of S-nitrosated mitochondrial proteins by S-nitrosothiol difference in gel electrophoresis (SNO-DIGE): implications for the regulation of mitochondrial function by reversible S-nitrosation. Biochem J. 2010;430(1):49-59.
- Lemasters JJ, Theruvath TP, Zhong Z, Nieminen AL. Mitochondrial calcium and the permeability transition in cell death. Biochimica et Biophysica Acta 2009;1787(11):1395–401.
- Nguyen TTM, Stevens M, Kohr M, Steenbergen C, Sack M, Murphy E. S-nitrosylation of cyclophilin D alters mitochondrial permeability transition pore. The FASEB Journal. 2011;25(1_supplement):1033.1-.1.
- Ohtani H, Katoh H, Tanaka T, Saotome M, Urushida T, Satoh H, Hayashi H. Effects of nitric oxide on mitochondrial permeability transition pore and thiol-mediated responses in cardiac myocytes. Nitric Oxide. 2012;26(2):95-101.
- Shoshan-Barmatz V, De Pinto V, Zweckstetter M, Raviv Z, Keinan N, Arbel N. VDAC, a multi-functional mitochondrial protein regulating cell life and death. Mol Aspects Med. 2010;31(3):227-85.
- Cheng Q, Sedlic F, Pravdic D, Bosnjak ZJ, Kwok WM. Biphasic effect of nitric oxide on the cardiac voltage-dependent anion channe. FEBS Lett. 2011;585(2):328-34.
- Crow MT, Mani K, Nam YJ, Kitsis RN. The mitochondrial death pathway and cardiac myocyte apoptosis. Circ Res. 2004;95(957-70).
- Gobeil FJ, Zhu T, Brault S, Geha A, Vazquez-Tello A, Fortier A, Barbaz D, Checchin D, Hou X, Nader M, Bkaily G, Gratton JP, Heveker N, Ribeiro-da-Silva A, Peri K, Bard H, Chorvatova A, D'Orléans-Juste P, Goetzl EJ, Chemtob S. Nitric oxide signaling via nuclearized endothelial nitric-oxide synthase modulates expression of the immediate early genes iNOS and mPGES-1. The Journal of biological chemistry. 2006;281(23):16058-67.
- Aquilano K, Baldelli S, Ciriolo MR. Nuclear recruitment of neuronal nitric-oxide synthase by α-syntrophin is crucial for the induction of mitochondrial biogenesis. The Journal of biological chemistry. 2014;289(1):365-78.
- Jones RJ, Jourd'heuil D, Salerno JC, Smith SM, Singer HA. iNOS regulation by calcium/calmodulin-dependent protein kinase II in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2007;292(6):H2634-42.
- Villanueva C, Giulivi C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol Med. 2010;49(3):307-16.
- Raghunath A, Sundarraj K, Nagarajan R, Arfuso F, Bian J, Kumar AP, Sethi G, Perumal E. Antioxidant response elements: Discovery, classes, regulation and potential applications. Redox Biol. 2018;17:297-314.
- Teixeira TM, da Costa DC, Resende AC, Soulage CO, Bezerra FF, Daleprane JB. Activation of Nrf2-Antioxidant Signaling by 1,25-Dihydroxycholecalciferol Prevents Leptin-Induced Oxidative Stress and Inflammation in Human Endothelial Cells. J Nutr. 2017;47(4):506-13.
- Tonelli C, Chio IIC, Tuveson DA. Transcriptional Regulation by Nrf2. Antioxidants & redox signaling. 2018;29(17):1727-45.
- Um HC, Jang JH, Kim DH, Lee C, Surh YJ. Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric oxide. 2011;25(2):161-8.
- Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxidants & redox signaling. 2013;18(4):386-99.
- Baldelli S, Ciriolo MR. Altered S-nitrosylation of p53 is responsible for impaired antioxidant response in skeletal muscle during aging. Aging (Albany NY). 2016;8(12):3450-67.
- Cherry AD, Suliman HB, Bartz RR, Piantadosi CA. Peroxisome proliferator-activated receptor γ co-activator 1-α as a critical co-activator of the murine hepatic oxidative stress response and mitochondrial biogenesis in Staphylococcus aureus sepsis. The Journal of biological chemistry. 2014;289(1):41-52.
- Schonhoff CM, Daou MC, Jones SN, Schiffer CA, Ross AH. Nitric oxide-mediated inhibition of Hdm2-p53 binding. Biochemistry. 2002;41(46):13570-4.
- Wardyn JD, Ponsford AH, Sanderson CM. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem Soc Trans. 2015;43:621-6.
- Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72(11):1493-50.
- Reynaert NL, Ckless K, Korn SH, Vos N, Guala AS, Wouters EF, van der Vliet A, Janssen-Heininger YM. Nitric oxide represses inhibitory kappaB kinase through S-nitrosylation. Proc Natl Acad Sci U S A. 2004;101(21):8945-50.
- Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, Moretti M, De Smaele E, Beg AA, Tergaonkar V, Chandel NS, Franzoso G. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 2011;13(10):1272-9.
- Semenza GL. Hydroxylation of HIF-1: oxygen sensing at the molecular level. Physiology. 2004;19(4):176-82.
- Brune B, Zhou J. The role of nitric oxide (NO) in stability regulation of hypoxia inducible factor-1α (HIF-1α). Curr Med Chem. 2003;10:845-55.
- Terry LJ, Shows EB, Wente SR. Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science. 2007;318:1412-6.
- Fornerod M, Ohno M, Yoshida M, Mattaj IW. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997;90:1051-60.
- Fu SC, Huang HC, Horton P, Juan HF. ValidNESs: a database of validated leucine-rich nuclear export signals. Nucleic Acids Res. 2013;41:D338-43.
- Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, Murli S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009;69(2):510-7.
- Wang P, Liu GH, Wu K, Qu J, Huang B, Zhang X, Zhou X, Gerace L, Chen C. Repression of classical nuclear export by S-nitrosylation of CRM1. J Cell Sci. 2009;122(Pt 20):3772-9.
- Freedman DA, Levine AJ. Nuclear export is required for degradation of endogenous p53 by MDM2 and human papillomavirus E6. Mol Cell Biol. 1998;18(12):7288-93.
- Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Proteomic analysis of S-nitrosylation and denitrosylation by resin-assisted capture. Nat Biotechnol. 2009;27(6):557-9.
- Azam S, Jouvet N, Jilani A, Vongsamphanh R, Yang X, Yang S, Ramotar D. Human glyceraldehyde-3-phosphate dehydrogenase plays a direct role in reactivating oxidized forms of the DNA repair enzyme APE1. J Bioil Chem. 2008;283(45):30632-41.
- Dobbs TA, Tainer JA, Lees-Miller SP. A structural model for regulation of NHEJ by DNA-PKcs autophosphorylation. DNA Repair (Amst). 201-;9(12):1307-14.
- Xu W, Liu L, Smith GC, Charles lG. Nitric oxide upregulates expression of DNA-PKcs to protect cells from DNA-damaging anti-tumour agents. Nat Cell Biol. 2000;2(6):339-45.
- Tang CH, Wei W, Liu L. Regulation of DNA repair by S-nitrosylation. Biochim Biophys Acta. 2012;1820(6):730-5.
- Tell G, Quadrifoglio F, Tiribelli C, Kelley MR. The many functions of APE1/Ref-1: not only a DNA repair enzyme. . Antioxid Redox Signal. 2009;11:601-20.
- Choi S, Joo HK, Jeon BH. Dynamic Regulation of APE1/Ref-1 as a Therapeutic Target Protein. Chonnam Med J. 2016;52(2):75-80.
- Qu J, Liu GH, Huang B, Chen C. Nitric oxide controls nuclear export of APE1/Ref-1 through S-nitrosation of cysteines 93 and 310. Nucleic Acids Res. 2007;35(8):2522-32.
- Ottaviano FG, Handy DE, Loscalzo J. Redox regulation in the extracellular environment. Circ J. 2008;72(1):1-16.
- Stamler JS, Lamas S, Fang FC. Nitrosylation. the prototypic redox-based signaling mechanism. Cell. 2001;106(6):675-83.
- Koh YH, Suzuki K, Che W, Park YS, Miyamoto Y, Higashiyama S, Taniguchi N. Inactivation of glutathione peroxidase by NO leads to the accumulation of H2O2 and the induction of HB-EGF via c-Jun NH2-terminal kinase in rat aortic smooth muscle cells. FASEB J. 2001;15:1472–4.
- Ortega-Galisteo AP, Rodríguez-Serrano M, Pazmiño DM, Gupta DK, Sandalio LM, Romero-Puertas MC. S-Nitrosylated proteins in pea (Pisum sativum L.) leaf peroxisomes: changes under abiotic stress. J Exp Bot. 2012;63(5):2089-103.
- Furuta S. Basal S-Nitrosylation Is the Guardian of Tissue Homeostasis. Trends Cancer. 2017;3(11):744-8. Epub 2017/11/10. doi: 10.1016/j.trecan.2017.09.003. PubMed PMID: 29120749.
- Subczynski WK, Lomnicka M, Hyde JS. Permeability of nitric oxide through lipid bilayer membranes. Free Radic Res. 1996;24(5):343-9.
- Herrera M, Hong NJ, Garvin JL. Aquaporin-1 transports NO across cell membranes. Hypertension. 2006;48(1):157-64.
- Denicola A, Souza JM, Radi R, Lissi E. Nitric oxide diffusion in membranes determined by fluorescence quenching. Arch Biochem Biophys. 1996;328(1):208-12.
- Malysheva EV, Kruglov SV, Khomenko IP, Bakhtina LY, Pshennikova MG, Manukhina EB, IY. M. Role of extracellular and intracellular nitric oxide in the regulation of macrophage responses. Bull Exp Biol Med. 2006;141(4):404-6.
- Matsumoto A, Gow AJ. Membrane transfer of S-nitrosothiols. Nitric Oxide. 2011;25(2):102-7.
- Iwakiri Y, Satoh A, Chatterjee S, Toomre DK, Chalouni CM, Fulton D, Groszmann R, Shah VH, Sessa WC. Nitric oxide synthase generates nitric oxide locally to regulate compartmentalized protein S-nitrosylation and protein trafficking. Proc Natl Acad Sci U S A. 2006;103(52):19777-82.
- Sangwung P, Greco TM, Wang Y, Ischiropoulos H, Sessa WC, Iwakiri Y. Proteomic identification of S-nitrosylated Golgi proteins: new insights into endothelial cell regulation by eNOS-derived NO. PLoS One. 2012;7(2):e31564.
- Bienert GP, Chaumont F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim Biophys Acta. 2014;1840(5):1596-604.
- Hess DT, Matsumoto A, Nudelman R, Stamler JS. S-nitrosylation: spectrum and specificity. Nature Cell Biology. 2001;3(2):E46-E8.
- Savage DF, Stroud RM. Structural basis of aquaporin inhibition by mercury. J Mol Biol. 2007;368(3):607-17.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245-313.
- Matono R, Miyano K, Kiyohara T, Sumimoto H. Arachidonic acid induces direct interaction of the p67(phox)-Rac complex with the phagocyte oxidase Nox2, leading to superoxide production. The Journal of biological chemistry. 2014;289(36):24874-84.
- Lou Z, Wang AP, Duan XM, Hu GH, Song GL, Zuo ML, Yang ZB. Upregulation of NOX2 and NOX4 Mediated by TGF-β Signaling Pathway Exacerbates Cerebral Ischemia/Reperfusion Oxidative Stress Injury. Cell Physiol Biochem. 2018;46(5):2103-13.
- Selemidis S, Dusting G, Peshavariya H, Kemp-Harper BK, Drummond GR. Nitric oxide suppresses NADPH oxidase-dependent superoxide production by S-nitrosylation in human endothelial cells. Cardiovasc Res. 2007;75(2):349-58.
- Harrison CB, Drummond GR, Sobey CG, Selemidis S. Evidence that nitric oxide inhibits vascular inflammation and superoxide production via a p47phox-dependent mechanism in mic. Clin Exp Pharmacol Physiol. 2010;37(4):429-34.
- Schmidt HM, Kelley EE, Straub AC. The impact of xanthine oxidase (XO) on hemolytic diseases. Redox Biol. 2019;21:101072.
- White CR. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proc Natl Acad Sci U S A. 1996;93(16):8745–9.
- Khan SA, Lee K, Minhas KM, Gonzalez DR, Raju SV, Tejani AD, Li D, Berkowitz DE, Hare JM. Neuronal nitric oxide synthase negatively regulates xanthine oxidoreductase inhibition of cardiac excitation-contraction coupling. Proc Natl Acad Sci U S A. 2004;101(45):15944-8.
- Saraiva RM, Minhas KM, Zheng M, Pitz E, Treuer A, Gonzalez D, Schuleri KH, Vandegaer KM, Barouch LA, Hare JM. Reduced neuronal nitric oxide synthase expression contributes to cardiac oxidative stress and nitroso-redox imbalance in ob/ob mice. Nitric oxide. 2007;16(3):331-8.
- Petersen SV, Oury TD, Ostergaard L, Valnickova Z, Wegrzyn J, IB T, C J, RP B, CL F, JD C, JJ. E. Extracellular superoxide dismutase (EC-SOD) binds to type i collagen and protects against oxidative fragmentation. The Journal of biological chemistry. 2004;279(14):13705-10.
- Jung O, Marklund SL, Geiger H, Pedrazzini T, Busse R, Brandes RP. Extracellular superoxide dismutase is a major determinant of nitric oxide bioavailability: in vivo and ex vivo evidence from ecSOD-deficient mice. Circ res. 2003;93(7):622-9.
- Holzmeister C, Gaupels F, Geerlof A, Sarioglu H, Sattler M, Durner J, Lindermayr C. Differential inhibition of Arabidopsis superoxide dismutases by peroxynitrite-mediated tyrosine nitration. J Exp Bot. 2015;66(3):989-99.
- Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017;11:613-9.
- Hervé-Grépinet V, Veillard F, Godat E, Heuzé-Vourc'h N, Lecaille F, Lalmanach G. Extracellular catalase activity protects cysteine cathepsins from inactivation by hydrogen peroxide. EFEBS Lett. 2008;582(9):1307-12.
- Lee JE, Kim H, Jang H, Cho EJ, Youn HD. Hydrogen peroxide triggers the proteolytic cleavage and the inactivation of calcineurin. J Neurochem. 2007;100(6):1703-12.
- Yoon SO, Park SJ, Yoon SY, Yun CH, Chung AS. Sustained production of H(2)O(2) activates pro-matrix metalloproteinase-2 through receptor tyrosine kinases/phosphatidylinositol 3-kinase/NF-kappa B pathway. The Journal of biological chemistry. 2002;277(33):30271-82.
- Parakh S, Atkin JD. Novel roles for protein disulphide isomerase in disease states: a double edged sword? Front Cell Dev Bil. 2015;3:30.
- Bekendam RH, Iyu D, Passam F, Stopa JD, De Ceunynck K, Muse O, Bendapudi PK, Garnier CL, Gopal S, Crescence L, Chiu J, Furie B, Panicot-Dubois L, Hogg PJ, Dubois C, Flaumenhaft R. Protein disulfide isomerase regulation by nitric oxide maintains vascular quiescence and controls thrombus formation. J Thromb Haemost. 2018;16(11):2322-35.
- Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein s-nitrosylation in neurodegenerative diseases. Neuron. 2013;78(4):596-614.
- Aranda E, Lopez-Pedrera C, De La Haba-Rodriguez JR, Rodriguez-Ariza A. Nitric oxide and cancer: the emerging role of S-nitrosylation. Curr Mol Med. 2012;12(1):50-67. Epub 2011/11/16. PubMed PMID: 22082481.
- Benhar M. Emerging Roles of Protein S-Nitrosylation in Macrophages and Cancer cells2016. doi: 10.2174/0929867323666160627.
- Balkwill FR, Capasso M, Hagemann T. The tumor microenvironment at a glance. J Cell Sci. 2012;125(Pt 23):5591-6. Epub 2013/02/20. doi: 10.1242/jcs.116392. PubMed PMID: 23420197.
- Jezierska-Drutel A, Rosenzweig SA, Neumann CA. Role of oxidative stress and the microenvironment in breast cancer development and progression. Adv Cancer Res. 2013;119:107-25. Epub 2013/07/23. doi: 10.1016/B978-0-12-407190-2.00003-4. PubMed PMID: 23870510; PMCID: PMC3950899.
- Wang M, Zhao J, Zhang L, Wei F, Lian Y, Wu Y, Gong Z, Zhang S, Zhou J, Cao K, Li X, Xiong W, Li G, Zeng Z, Guo C. Role of tumor microenvironment in tumorigenesis. J Cancer. 2017;8(5):761-73. Epub 2017/04/07. doi: 10.7150/jca.17648. PubMed PMID: 28382138; PMCID: PMC5381164.
- Cook JA, Gius D, Wink DA, Krishna MC, Russo A, Mitchell JB. Oxidative stress, redox, and the tumor microenvironment. Semin Radiat Oncol. 2004;14(3):259-66. Epub 2004/07/16. doi: 10.1016/j.semradonc.2004.04.001. PubMed PMID: 15254869.
- Fiaschi T, Chiarugi P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: a diabolic liaison. Int J Cell Biol. 2012;2012:762825. Epub 2012/06/06. doi: 10.1155/2012/762825. PubMed PMID: 22666258; PMCID: PMC3361160.
- Ray PD, Huang BW, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. 2012;24(5):981-90. Epub 2012/01/31. doi: 10.1016/j.cellsig.2012.01.008. PubMed PMID: 22286106; PMCID: PMC3454471.
- BeleÂn Beltrain, Antonia Orsi, Emilio Clementi, Moncada S. Oxidative stress and S Nitrosylation of proteins in cells2000.
- Lorenzen I, Mullen L, Bekeschus S, Hanschmann EM. Redox Regulation of Inflammatory Processes Is Enzymatically Controlled. Oxid Med Cell Longev. 2017;2017:8459402. Epub 2017/11/10. doi: 10.1155/2017/8459402. PubMed PMID: 29118897; PMCID: PMC5651112.
- Noel A, Jost M, Maquoi E. Matrix metalloproteinases at cancer tumor-host interface. Semin Cell Dev Biol. 2008;19(1):52-60. Epub 2007/07/13. doi: 10.1016/j.semcdb.2007.05.011. PubMed PMID: 17625931.
- McCarthy SM, Bove PF, Matthews DE, Akaike T, van der Vliet A. Nitric oxide regulation of MMP-9 activation and its relationship to modifications of the cysteine switch. Biochemistry. 2008;47(21):5832-40. Epub 2008/05/03. doi: 10.1021/bi702496v. PubMed PMID: 18452312; PMCID: PMC3030771.
- Reis ST, Leite KRM, Piovesan LF, Pontes-Junior J, Viana NI, Abe DK, Crippa A, Moura CM, Adonias SP, Srougi M, Dall'Oglio MF. Increased expression of MMP-9 and IL-8 are correlated with poor prognosis of Bladder Cancer. BMC Urol. 2012;12:18-. doi: 10.1186/1471-2490-12-18. PubMed PMID: 22695075.
- Mercedes Ferrerasa, Ute Felborb, Thomas Lenharda, Bjorn R. Olsen, Delaisse J-M. Generation and degradation of human endostatin proteins by various proteinases2000.
- Harris LK, McCormick J, Cartwright JE, Whitley GS, Dash PR. S-nitrosylation of proteins at the leading edge of migrating trophoblasts by inducible nitric oxide synthase promotes trophoblast invasion. Exp Cell Res. 2008;314(8):1765-76. Epub 2008/04/09. doi: 10.1016/j.yexcr.2008.02.010. PubMed PMID: 18394602.
- Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GV, Mehta K. Transglutaminase regulation of cell function. Physiol Rev. 2014;94(2):383-417. Epub 2014/04/03. doi: 10.1152/physrev.00019.2013. PubMed PMID: 24692352; PMCID: PMC4044299.
- Lai TS, Hausladen A, Slaughter TF, Eu JP, Stamler JS, Greenberg CS. Calcium regulates S-nitrosylation, denitrosylation, and activity of tissue transglutaminase. Biochemistry. 2001;40(16):4904-10. Epub 2001/04/18. doi: 10.1021/bi002321t. PubMed PMID: 11305905.
- Huang L, Xu AM, Liu W. Transglutaminase 2 in cancer. Am J Cancer Res. 2015;5(9):2756-76. PubMed PMID: 26609482.
- Mangala LS, Mehta K. Tissue transglutaminase (TG2) in cancer biology. Prog Exp Tumor Res. 2005;38:125-38. Epub 2005/03/05. doi: 10.1159/000084237. PubMed PMID: 15746533.
- Fraser M, Chan SL, Chan SS, Fiscus RR, Tsang BK. Regulation of p53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene. 2006;25(15):2203-12
- Postovit LM. Role of Nitric Oxide in the Regulation of the Pro-tumourigenic Hypoxic Phenotype: From Instigation to Mitigation. In: Bonavida B, editor. Nitric oxide and Cancer: Pathogenesis and Therapy: Springer Publishing; 2015. p. 65-84.
- Nath N, Kashfi K, Chen J, Rigas B. Nitric oxide-donating aspirin inhibits β-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear β-catenin–TCF association. Proc Natl Acad Sci U S A. 2003;100(22):12584-9. doi: 10.1073/pnas.2134840100.
- Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, Power GG, Kelm M, Gladwin MT, Schechter AN. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106(2):734-9.
This entry is adapted from the peer-reviewed paper 10.3390/antiox8090404