 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Siyer Roohani | + 3118 word(s) | 3118 | 2021-07-15 05:09:03 | | | |
| 2 | Peter Tang | Meta information modification | 3118 | 2021-07-29 03:31:03 | | |
Video Upload Options
The liver is an essential immunological organ due to its gatekeeper position to bypassing antigens from the intestinal blood flow and microbial products from the intestinal commensals. The tissue-resident liver macrophages, termed Kupffer cells, represent key phagocytes that closely interact with local parenchymal, interstitial and other immunological cells in the liver to maintain homeostasis and tolerance against harmless antigens. Upon liver injury, the pool of hepatic macrophages expands dramatically by infiltrating bone marrow-/monocyte-derived macrophages. The interplay of the injured microenvironment and altered macrophage pool skews the subsequent course of liver injuries. It may range from complete recovery to chronic inflammation, fibrosis, cirrhosis and eventually hepatocellular cancer.
1. Introduction
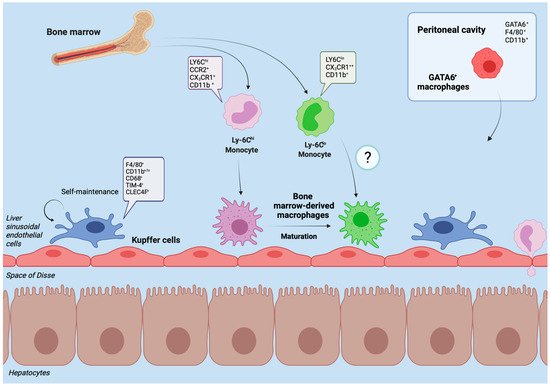
2. Hepatic Macrophages in Homeostasis
3. Hepatic Macrophages in Acute Liver Injury
4. Chronic Liver Injury
4.1. Hepatic Macrophages in Nonalcoholic and Alcoholic Fatty Liver Disease
4.2. Hepatic Macrophages in Viral Hepatitis B and C
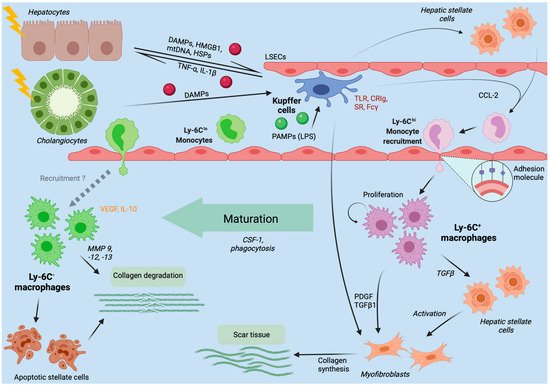
4.3. Fibrosis and Cirrhosis Modulation by Hepatic Macrophages
5. Therapeutic Approaches
References
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321.
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186.
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684.
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56.
- Abdullah, Z.; Knolle, P.A. Liver macrophages in healthy and diseased liver. Pflugers Arch. 2017, 469, 553–560.
- Gola, A.; Dorrington, M.G.; Speranza, E.; Sala, C.; Shih, R.M.; Radtke, A.J.; Wong, H.S.; Baptista, A.P.; Hernandez, J.M.; Castellani, G.; et al. Commensal-driven immune zonation of the liver promotes host defence. Nature 2021, 589, 131–136.
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566.
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312.
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015, 62, 279–291.
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321.
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol. 2014, 193, 344–353.
- von Kupffer, K. Über Sternzellen der Leber. Arch. Mikroskop Anat. 1876, 12, 353–358.
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435.
- Horst, A.K.; Tiegs, G.; Diehl, L. Contribution of Macrophage Efferocytosis to Liver Homeostasis and Disease. Front. Immunol. 2019, 10, 2670.
- Soucie, E.L.; Weng, Z.; Geirsdottir, L.; Molawi, K.; Maurizio, J.; Fenouil, R.; Mossadegh-Keller, N.; Gimenez, G.; VanHille, L.; Beniazza, M.; et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 2016, 351, aad5510.
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9.
- Guilliams, M.; Scott, C.L. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 2017, 17, 451–460.
- David, B.A.; Rezende, R.M.; Antunes, M.M.; Santos, M.M.; Freitas Lopes, M.A.; Diniz, A.B.; Sousa Pereira, R.V.; Marchesi, S.C.; Alvarenga, D.M.; Nakagaki, B.N.; et al. Combination of Mass Cytometry and Imaging Analysis Reveals Origin, Location, and Functional Repopulation of Liver Myeloid Cells in Mice. Gastroenterology 2016, 151, 1176–1191.
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and tissue specialization. Annu. Rev. Immunol. 2015, 33, 643–675.
- Strnad, P.; Tacke, F.; Koch, A.; Trautwein, C. Liver—Guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 55–66.
- Terpstra, V.; van Berkel, T.J. Scavenger receptors on liver Kupffer cells mediate the in vivo uptake of oxidatively damaged red blood cells in mice. Blood 2000, 95, 2157–2163.
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.; Holderried, T.A.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951.
- Scott, C.L.; Guilliams, M. The role of Kupffer cells in hepatic iron and lipid metabolism. J. Hepatol. 2018, 69, 1197–1199.
- Kanamori, Y.; Tanaka, M.; Itoh, M.; Ochi, K.; Ito, A.; Hidaka, I.; Sakaida, I.; Ogawa, Y.; Suganami, T. Iron-rich Kupffer cells exhibit phenotypic changes during the development of liver fibrosis in NASH. iScience 2021, 24, 102032.
- van Vliet, S.J.; Gringhuis, S.I.; Geijtenbeek, T.B.H.; van Kooyk, Y. Regulation of effector T cells by antigen-presenting cells via interaction of the C-type lectin MGL with CD45. Nat. Immunol. 2006, 7, 1200–1208.
- Deppermann, C.; Kratofil, R.M.; Peiseler, M.; David, B.A.; Zindel, J.; Castanheira, F.; van der Wal, F.; Carestia, A.; Jenne, C.N.; Marth, J.D.; et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 2020, 217, e20190723.
- Wang, Y.; van der Tuin, S.; Tjeerdema, N.; van Dam, A.D.; Rensen, S.S.; Hendrikx, T.; Berbée, J.F.; Atanasovska, B.; Fu, J.; Hoekstra, M.; et al. Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 2015, 62, 1710–1722.
- Thompson, A.; Di Angelantonio, E.; Sarwar, N.; Erqou, S.; Saleheen, D.; Dullaart, R.P.; Keavney, B.; Ye, Z.; Danesh, J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA 2008, 299, 2777–2788.
- van der Tuin, S.J.L.; Li, Z.; Berbee, J.F.P.; Verkouter, I.; Ringnalda, L.E.; Neele, A.E.; van Klinken, J.B.; Rensen, S.S.; Fu, J.; de Winther, M.P.J.; et al. Lipopolysaccharide Lowers Cholesteryl Ester Transfer Protein by Activating F4/80(+)Clec4f(+)Vsig4(+)Ly6C(-) Kupffer Cell Subsets. J. Am. Heart Assoc. 2018, 7, e008105.
- European Association for the Study of the Liver; Wendon, J.; Panel, M.; Cordoba, J.; Dhawan, A.; Larsen, F.S.; Manns, M.; Samuel, D.; Simpson, K.J. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure. J. Hepatol. 2017, 66, 1047–1081.
- Bernal, W.; Wendon, J. Acute Liver Failure. N. Engl. J. Med. 2013, 369, 2525–2534.
- Hadem, J.; Tacke, F.; Bruns, T.; Langgartner, J.; Strnad, P.; Denk, G.U.; Fikatas, P.; Manns, M.P.; Hofmann, W.P.; Gerken, G.; et al. Etiologies and outcomes of acute liver failure in Germany. Clin. Gastroenterol. Hepatol. 2012, 10, 664–669.e2.
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immun. Netw. 2018, 18, e27.
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104.
- Weiskirchen, R.; Tacke, F. Liver Fibrosis: From Pathogenesis to Novel Therapies. Dig. Dis. 2016, 34, 410–422.
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Karlsen, T.H.; Tacke, F. The times they are a’changin—Positioning the European Association for the Study of the Liver in the changing landscape of hepatology. J. Hepatol. 2018, 68, 873–875.
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133.
- Di Cesare, M.; Sorić, M.; Bovet, P.; Miranda, J.J.; Bhutta, Z.; Stevens, G.A.; Laxmaiah, A.; Kengne, A.-P.; Bentham, J. The epidemiological burden of obesity in childhood: A worldwide epidemic requiring urgent action. BMC Med. 2019, 17, 212.
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402.
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagström, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wai-Sun Wong, V.; Peleg, N.; et al. Association Between Fibrosis Stage and Outcomes of Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625.e12.
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565.
- Povsic, M.; Wong, O.Y.; Perry, R.; Bottomley, J. A Structured Literature Review of the Epidemiology and Disease Burden of Non-Alcoholic Steatohepatitis (NASH). Adv. Ther. 2019, 36, 1574–1594.
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.e17.
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64.
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965.
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver. Dis. 2020, 40, 298–306.
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016, 150, 956–967.
- Bartneck, M.; Fech, V.; Ehling, J.; Govaere, O.; Warzecha, K.T.; Hittatiya, K.; Vucur, M.; Gautheron, J.; Luedde, T.; Trautwein, C.; et al. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology 2016, 63, 1310–1324.
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization through Downregulation of PlGF. Cancer Cell 2011, 19, 31–44.
- Bereshchenko, O.; Migliorati, G.; Bruscoli, S.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper: A Novel Anti-inflammatory Molecule. Front. Pharmacol. 2019, 10, 308.
- Robert, O.; Boujedidi, H.; Bigorgne, A.; Ferrere, G.; Voican, C.S.; Vettorazzi, S.; Tuckermann, J.P.; Bouchet-Delbos, L.; Tran, T.; Hemon, P.; et al. Decreased expression of the glucocorticoid receptor-GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J. Hepatol. 2016, 64, 916–924.
- Wiktor, S.Z.; Hutin, Y.J. The global burden of viral hepatitis: Better estimates to guide hepatitis elimination efforts. Lancet 2016, 388, 1030–1031.
- Vercauteren, K.; de Jong, Y.P.; Meuleman, P. HCV animal models and liver disease. J. Hepatol. 2014, 61, S26–S33.
- Movita, D.; van de Garde, M.D.; Biesta, P.; Kreefft, K.; Haagmans, B.; Zuniga, E.; Herschke, F.; De Jonghe, S.; Janssen, H.L.; Gama, L.; et al. Inflammatory monocytes recruited to the liver within 24 h after virus-induced inflammation resemble Kupffer cells but are functionally distinct. J. Virol. 2015, 89, 4809–4817.
- Boltjes, A.; van Montfoort, N.; Biesta, P.J.; Op den Brouw, M.L.; Kwekkeboom, J.; van der Laan, L.J.W.; Janssen, H.L.A.; Boonstra, A.; Woltman, A.M. Kupffer Cells Interact With Hepatitis B Surface Antigen In Vivo and In Vitro, Leading to Proinflammatory Cytokine Production and Natural Killer Cell Function. J. Infect. Dis. 2014, 211, 1268–1278.
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782.
- Shrivastava, S.; Mukherjee, A.; Ray, R.; Ray, R.B. Hepatitis C virus induces interleukin-1β (IL-1β)/IL-18 in circulatory and resident liver macrophages. J. Virol. 2013, 87, 12284–12290.
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487.
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330.
- Frith, J.C.; Mönkkönen, J.; Blackburn, G.M.; Russell, R.G.G.; Rogers, M.J. Clodronate and Liposome-Encapsulated Clodronate Are Metabolized to a Toxic ATP Analog, Adenosine 5′-(β,γ-Dichloromethylene) Triphosphate, by Mammalian Cells In Vitro. J. Bone Miner. Res. 1997, 12, 1358–1367.
- Lang, P.A.; Recher, M.; Honke, N.; Scheu, S.; Borkens, S.; Gailus, N.; Krings, C.; Meryk, A.; Kulawik, A.; Cervantes-Barragan, L.; et al. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 2010, 52, 25–32.
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Aspects Med. 2019, 65, 2–15.
- Maurice, J.; Pinzani, M. The stratification of cirrhosis. Hepatol. Res. 2020, 50, 535–541.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460.
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729.
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411.
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65.
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110.
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol. Life Sci. 2016, 73, 1969–1987.
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e7.
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147.
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473.
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757.
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J. Immunol. 2013, 190, 5226–5236.
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195.
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072.
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274.
- Liu, Y.; Munker, S.; Müllenbach, R.; Weng, H.-L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116.
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.K.; et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398.
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665.
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337.
- Campana, L.; Starkey Lewis, P.J.; Pellicoro, A.; Aucott, R.L.; Man, J.; O’Duibhir, E.; Mok, S.E.; Ferreira-Gonzalez, S.; Livingstone, E.; Greenhalgh, S.N.; et al. The STAT3–IL-10–IL-6 Pathway Is a Novel Regulator of Macrophage Efferocytosis and Phenotypic Conversion in Sterile Liver Injury. J. Immunol. 2018, 200, 1169–1187.
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076.
- Lambrecht, J.; van Grunsven, L.A.; Tacke, F. Current and emerging pharmacotherapeutic interventions for the treatment of liver fibrosis. Expert Opin. Pharmacother. 2020, 21, 1637–1650.
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat. Med. 2019, 25, 1560–1565.
- Starkey Lewis, P.J.; Moroni, F.; Forbes, S.J. Macrophages as a Cell-Based Therapy for Liver Disease. Semin. Liver. Dis. 2019, 39, 442–451.

