The liver is an essential immunological organ due to its gatekeeper position to bypassing antigens from the intestinal blood flow and microbial products from the intestinal commensals. The tissue-resident liver macrophages, termed Kupffer cells, represent key phagocytes that closely interact with local parenchymal, interstitial and other immunological cells in the liver to maintain homeostasis and tolerance against harmless antigens. Upon liver injury, the pool of hepatic macrophages expands dramatically by infiltrating bone marrow-/monocyte-derived macrophages. The interplay of the injured microenvironment and altered macrophage pool skews the subsequent course of liver injuries. It may range from complete recovery to chronic inflammation, fibrosis, cirrhosis and eventually hepatocellular cancer.
1. Introduction
Apart from its central role as a metabolic organ, the liver is of critical importance for immune homeostasis and immunological responses to injury. It is located at an intersection of systemic circulation receiving arterial blood through the hepatic artery and portal venous gut-derived blood, enriched with nutrients and microbial products. This gatekeeper position gives the liver not only the challenging task to extract and adapt to innocuous nutrient antigens, but also to identify, bind, inactivate and filter potentially harmful antigens before entering the systemic circulation. To fulfill these tasks the liver harbors the largest population of tissue macrophages in the human body, the so-called Kupffer cells [
1,
2,
3]. Hepatic macrophages consist of tissue-resident Kupffer cells and bone marrow-derived monocytes migrating from the blood stream into the liver tissue, giving rise to bone marrow-derived macrophages [
4]. Within the fine microanatomical meshwork of sinusoidal vessels supplying each hepatocyte (the functional, metabolic unit in the liver) Kupffer cells are commonly situated within two branching sinusoids in the periportal area. Equipped with long cytoplasmic protrusions located close to the entrance of supplying blood vessels, these immobile, self-sustaining and locally proliferating macrophages specialize in clearance and gatekeeper function to incoming antigens [
5]. Interestingly, most recent studies revealed the asymmetric localization of immune cells in the liver to be orchestrated by liver sinusoidal endothelial cells (LSECs) and adapted to incoming commensal bacteria. LSECs sense commensal bacteria, adjust the pericellular matrix and generate chemokine gradients for immune cells to form zones in order to optimize host defense [
6].
In contrast to Kupffer cells, bone marrow-derived macrophages account for only a minor proportion of the hepatic macrophage pool in the healthy liver; they are mainly located at the portal triad [
1]. Principally, Kupffer cells are considered tolerogenic phagocytes while bone marrow-derived, infiltrating macrophages represent an inflammatory phenotype. However, the functional phenotype and differentiation is highly diverse and dependent on the interaction between these phagocytes and their microenvironment. Therefore, the traditionally applied model of M1 (inflammatory) and M2 (anti-inflammatory) macrophages [
7], which is based on “artificially” induced polarization states in vitro, does not adequately reflect the complex heterogeneity of macrophage polarization in the liver in vivo [
8].
Upon liver injury, bone marrow-derived monocytes rapidly infiltrate the liver tissue and differentiate into macrophages, thereby largely augmenting the hepatic macrophage pool. In damaged liver, the microenvironment alters radically. Inflammatory mediators are released by activated and stressed cells while the number of dead cells increases. These microenvironmental changes have a tremendous impact on the phenotypes of Kupffer cells and monocyte-derived macrophages. Their phenotypes determine the functional contribution of macrophages to tissue restoration or aggravation of liver injury [
9,
10,
11].
2. Hepatic Macrophages in Homeostasis
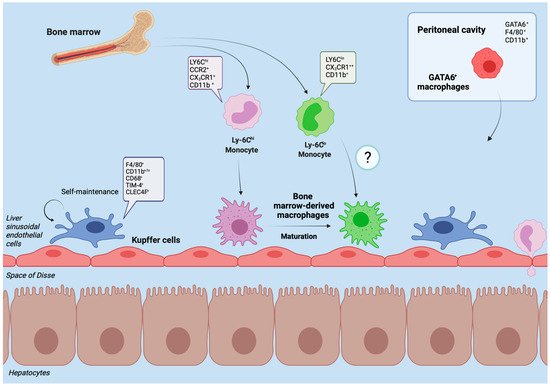
Under homeostatic conditions Kupffer cells, named after the German anatomist Karl Wilhelm von Kupffer (1829–1902) [
12], are the predominate macrophage population in the liver. The initial notion that these non-migratory, tissue-resident, and self-sustaining phagocytes embryonically originate from hematopoietic stem cells [
13] was recently challenged. Current data suggests that Kupffer cells derive from yolk sac erythromyeloid progenitors that colonize the fetal liver in embryonic day 8.5 in mice. At embryonic day 9.5 these cells give rise to macrophage precursors via signaling of the CX
3C chemokine receptor 1 (CX
3CR-1). Through upregulation of the transcription factor inhibitor of DNA binding 3 (ID3) the macrophage precursors eventually develop into Kupffer cells [
14]. The half-life of a Kupffer cell is 12.4 days in mice, replenishment occurs through self-renewal which is tightly regulated by the transcription factors MafB and c-Maf [
15]. Nonetheless, in case of Kupffer cell death or depletion, as it occurs in acute liver injury, circulating monocytes can contribute to the Kupffer cell pool as well [
16] (
Figure 1). This adaptability suggests that monocytes as hematopoietic derivates may develop into Kupffer Cells despite their distinct embryonic origin [
17,
18].
Figure 1. Liver macrophages in homeostasis. The figure displays the heterogenous subpopulations of macrophages in the healthy liver of mouse models. Kupffer cells represent the self-renewing, tissue-resident and dominant phagocyte population under healthy conditions. They reside immovably along the sinusoids, close to the entrance of supplying blood vessels to effectively sort out and process bypassing, gut-derived antigens and microbial products. Through close contact with parenchymal cells and the sinusoidal blood flow, Kupffer cells act as sensors of tissue integrity and gatekeepers for initiating or suppressing immune responses. In the adult liver of healthy mice, a minor proportion of macrophages derives from infiltrating monocytes (originating from the bone marrow). Upon liver injury, their number may rapidly expand. In mice, infiltrating Ly-6Chi macrophages initiate inflammation while circulating Ly-6Clo macrophages have a more mature phenotype, patrolling through the liver and promoting tissue restoration. After infiltration, Ly-6Chi macrophages can mature to Ly-6Clo macrophages. The peritoneal cavity contains GATA6+ macrophages that can promptly cross the mesothelium and infiltrate into the subcapsular area of the liver to support tissue repair. Typical markers of the different macrophage populations in mouse models are indicated.
Kupffer cells have numerous essential functions for tissue integrity far beyond being highly effective phagocytes. Apart from the recognition, ingestion and processing of foreign material and cellular debris [
19], Kupffer cells play a key role in antimicrobial defense [
20]. Equally important is their mediator function in maintaining tolerance to innocuous antigens that continuously pass through the liver before entering the systemic circulation [
9].
Moreover, Kupffer cells participate in important metabolic pathways. They recycle iron by phagocytosing damaged red blood cells (RBC) and metabolizing heme-derived, histotoxic free iron [
21,
22,
23]. Recent evidence suggests that hepatic iron accumulation, particularly heavily iron-loaded Kupffer cells, are involved in the pathogenesis of nonalcoholic steatohepatitis (NASH) and liver fibrosis [
24]. Kupffer cells are also involved in the degradation of aged platelets, as the depletion of Kupffer cells has prevented the removal of aged platelets on intravital imaging. Thereby, the calcium-dependent c-type lectin named macrophage galactose lectin (MGL) receptor on macrophages [
25] interacts with desialylated, aged and potentially dysfunctional platelets [
26].
Additionally, resting Kupffer cells are the main source of the plasma level of cholesteryl ester transfer protein (CETP) [
27]. This pore-forming protein enables the transfer of cholesterol from the antiatherogenic, anti-inflammatory high-density lipoproteins (HDL) to the atherogenic low (LDL) and very low-density lipoproteins (VLDL), that deliver cholesterol to the peripheral tissue [
28]. Interestingly, lipopolysaccharides (LPS) from Gram-negative bacteria induce Kupffer cell activation, which significantly reduces CETP synthesis, thereby raising HDL levels. These findings may imply that HDL is involved in host defense and that CETP and HDL signaling does not only correlate but is mechanistically connected to liver inflammation [
29].
3. Hepatic Macrophages in Acute Liver Injury
Acute liver injury is defined clinically as a (more than) two- to threefold increase of liver transaminases above the upper limit of normal (marker of hepatocyte damage), jaundice and impaired coagulation function (International Normalized Ratio (INR) > 1.5) of hepatic origin in a patient without preexisting chronic liver disease. Acute liver failure additionally comprises altered mentation (termed hepatic encephalopathy) and is initiated by a severe acute liver injury [
40], a life-threatening condition. The most common causes include hepatotoxic drugs (e.g., acetaminophen/paracetamol, phenprocoumon, antibiotics, antiepileptics), herbal or dietary supplements and acute viral hepatitis (hepatitis A (HAV), B (HBV) and E (HEV) viruses), although a multicenter data analysis from Germany revealed that in about one quarter of cases the cause remains unknown. Approximately one third to half of all patients with acute liver failure require emergency liver transplantation as a last resort [
41,
42].
The immense injury to the liver tissue greatly alters both, the microenvironment and immune cells. Danger-associated molecular patterns (DAMPs), a group of endogenous danger molecules released from damaged or dying cells such as high mobility group box 1 (HMGB1) or mitochondrial DNA (mtDNA) [
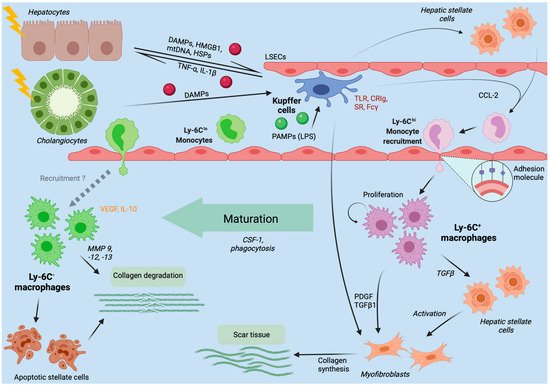
43] are released and bind to pattern recognition receptors (PRR) such as TLRs on Kupffer cells, thereby activating them (
Figure 2) [
44]. The recognition of these danger signals leads to the formation of the inflammasome.
Figure 2. Liver macrophages in the initiation, progression and regression of liver injury. The figure summarizes the role of hepatic macrophages in the course of liver injuries, as derived from mouse models. Kupffer cells immovably reside at the luminal side of liver sinusoidal endothelial cells (LSECs). Upon injury to hepatocytes or cholangiocytes, intracellular components like mitochondrial DNA (mtDNA) or heat shock proteins (HSP), functioning as danger-associated molecular patterns (DAMPs), are released. Moreover, microbial products (e.g., LPS) from the intestinal microbiota enter the liver via the portal venous blood flow and function as pathogen-associated molecular patterns (PAMPs). DAMPs and PAMPs activate Kupffer cells, which in turn secrete inflammatory cytokines (e.g., TNF-α, IL-1β) and CCL-2. While TNF-α and IL-1β further contribute to hepatocyte injury, CCL-2 recruits Ly-6Chi monocytes from the bloodstream to infiltrate the tissue and differentiate into inflammatory, fibrogenic and angiogenic Ly-6C+ macrophages. If the inflammation does not subside, Ly-6C+ macrophages will activate hepatic stellate cells (HSCs) to become collagen-producing myofibroblasts forming scare tissue. In case of inflammation resolution, Ly-6C+ macrophages will mature to restorative, anti-inflammatory Ly-6C− macrophages, that promote degradation of scar tissue by matrix degrading metalloproteinases (MMPs).
4. Chronic Liver Injury
4.1. Hepatic Macrophages in Nonalcoholic and Alcoholic Fatty Liver Disease
All forms of chronic liver injury, such as nonalcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD) or chronic viral hepatitis B and C are driven by continuous hepatic inflammation and lead to fibrosis as a uniform wound healing response. If the inflammatory status does not subside, fibrosis eventually leads to cirrhosis and hepatocellular carcinoma [
1,
80].
Nonalcoholic fatty liver disease is the most common liver disease affecting around 24% of the population worldwide and represents the liver manifestation of obesity and metabolic syndrome [
81]. Through tremendous advances in the treatment of viral hepatitis B and C on the one hand and the ever-increasing global burden of obesity even in younger generations on the other hand, NAFLD and its sequelae are estimated to become the most common causes of liver cirrhosis and hepatocellular carcinoma [
82,
83,
84].
NAFLD is an umbrella term for a spectrum of liver diseases in a patient with no other causes for secondary hepatic fat accumulation and particularly with no significant alcohol intake (<30 g/d for men, <20 g/d for women) [
85]. The typical pathophysiological sequence starts with hepatic steatosis, a histological accumulation of triglycerides in >5% of hepatocytes, to nonalcoholic steatohepatitis, a chronic inflammatory condition with necrotic hepatocyte injury leading to NASH-fibrosis, cirrhosis and eventually hepatocellular carcinoma [
85]. Yet, this process may not be considered linear as steatosis may also directly lead to fibrosis or inflammation resolution may improve and restore tissue integrity. The degree of fibrosis is defined histologically by the number of fibrotic septa from F0 (no fibrosis) over F1 (portal fibrosis without septa), F2 (portal fibrosis with few septa), F3 (numerous septa, “bridge building fibrosis”) to F4 being cirrhosis. The fibrotic stage is a predictor for liver-related morbidity, liver-related mortality and overall mortality [
86,
87]. In noncirrhotic NAFLD fibrosis (F0-F3 fibrosis) the main causes of death in NAFLD patients are cardiovascular diseases and non-hepatic malignancies [
88]. However, the liver-related mortality exponentially grows and supersedes all other causes of mortality once a cirrhotic liver condition (F4 fibrosis) is established [
89]. Nevertheless, NAFLD has to be considered the hepatic manifestation of a systemic burden which emphasizes the necessity for interdisciplinary care of patients with NAFLD [
90].
The fat overload in NAFLD and ALD causes cell death of hepatocytes and the release of DAMPs that activate macrophages and their inflammasomes [
91,
92]. Another inflammatory pathway was described in in vitro studies of NASH where fatty acids (palmitate or lysophosphatidyl-choline) were able to induce the release of extracellular vesicles containing TNF-α-related apoptosis-inducing ligands (TRAIL) from hepatocytes. The TRAILs increased the transcription of IL-1β and IL-6 in monocyte-derived macrophages from mice thereby promoting inflammation [
93]. Furthermore, the liver-derived plasma protein histidine-rich glycoprotein (HRP) has been shown to skew macrophages towards inflammatory phenotypes in mouse models of NASH and tumors [
94,
95]. The glucocorticoid-induced leucine zipper (GILZ), a multifunctional protein that spreads the anti-inflammatory signaling of glucocorticoids in numerous tissues, is downregulated in Kupffer cells of murine models of hepatic steatosis [
96]. Consequently, Kupffer cells displayed an inflammatory phenotype with elevated expression of CCL-2, TNF-α and IL-6, which was improved in GILZ overexpressing transgenic mice [
97].
4.2. Hepatic Macrophages in Viral Hepatitis B and C
Chronic viral hepatitis caused by HBV or HCV remain a meaningful cause of liver associated morbidity and mortality [
107]. The research on immunocompetent animal models is limited by the number of appropriate models as, for example, mice have a natural immunity against HCV and research on chimpanzees is hampered by financial and ethical constraints [
108].
Similar to their multifaceted tasks in other liver diseases, hepatic macrophages can contribute to antiviral responses upon HBV or HCV infection. Research on the hepatotropic lymphocytic chroriomeningitis virus (LCMV) in mice has shown that the liver rapidly recruits pro-inflammatory macrophages (within 24 h) to support local Kupffer cells, when acutely infected [
109]. In vitro cell culture studies of human Kupffer cells exposed to HBV surface antigen (HBsAg) have revealed increasing productions of the inflammatory cytokines TNF-α, IL-6 and CXCL8 (IL-8) that peaked after six hours of exposure [
110]. Through NF-κB mediated transcription, these inflammatory cytokines, most importantly, IL-6 prohibit viral spreading in infected hepatocytes. IL-6 activates the mitogen-activated protein kinases exogenous signal-regulated kinase 1/2 (ERK1/2), and c-jun N-terminal kinase (JNK), two members of the mitogen-activated protein kinases (MAPKs) that transfer extracellular stimuli to a wide range of cellular stimuli [
111]. ERK1/2 and JNK inhibit expression of hepatocyte nuclear factor (HNF) 1α and HNF4α, two transcription factors essential for HBV gene expression and replication [
112]. Similarly, human Kupffer cells and monocyte-derived macrophages incubated with HCV in vitro activated the inflammasome and NF-κB via TLR2, which induced IL-1β and IL-18 secretion [
113,
114]. In line with the aforementioned two signal activation of inflammasomes, HCV exposed human macrophages showed: (i) viral RNA triggers MyD88-mediated TLR7 signaling to induce IL-1β mRNA expression; (ii) HCV uptake concomitantly induces a potassium efflux that activates the NLRP3 inflammasome for IL-1β processing and secretion; (iii) HCV infection is directly linked to liver inflammation by NLRP3 inflammasome activation [
115].
Moreover, the depletion of Kupffer cells via clodronate-liposomes, a specific bisphosphonate that depletes macrophages [
116], led to a rapid dissemination of LCMV in mice due to the inability to capture and process viral particles [
117]. It is therefore appropriate to consider Kupffer cells a critical immune barrier in acute HBV and HCV infections.
4.3. Fibrosis and Cirrhosis Modulation by Hepatic Macrophages
Fibrosis describes excessive scarring overproportionate to a wound healing response towards tissue injury [
127]. If left untreated, progressive hepatic fibrosis leads to cirrhosis, a histoarchitectural remodeling with abundant collagen deposition that becomes clinically overt by decreasing hepatic function [
128]. The chronic inflammation predisposes to carcinogenesis while increasing portal pressure triggers numerous clinical complications with significant morbidity and mortality [
129]. Both, clinical observations and experimental models over the last years, challenged the former belief of irreversible liver fibrosis and identified new targets to halt and reverse this process [
130].
Hepatic stellate cells (HSCs) transition to myofibroblasts and represent the main matrix-producing cells in the liver. Activation is induced by direct cell–cell interaction or binding of fibrogenic mediators [
131].
In the dynamic process of inflammation and fibrogenesis, liver macrophages hold a dual function; during fibrogenesis the depletion of hepatic macrophages in mice improves scarring, whereas the depletion during resolution phases impedes adequate tissue restoration pointing towards functionally distinct subpopulations of macrophages within this process [
132]. Upon damage to the hepatic microenvironment DAMPs and PAMPs are released that trigger local non-parenchymal cells (Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells) to release a broad variety of inflammatory and profibrogenic soluble mediators. These mediators (e.g., CCL2) attract inflammatory immune cells like LyC6
hi bone marrow-derived macrophages and activate matrix-producing profibrotic cell populations to form scar tissue [
34,
127]. Besides the recruitment of inflammatory and profibrotic macrophages by CCL2, Kupffer cells can also directly promote activation and survival of HSCs and myofibroblasts through secretion of growth factors (PDGF, TGF-β) and CCL5 [
133,
134,
135,
136,
137].
The interaction of Kupffer cells with other immune cells, such as natural killer T (NKT) cells, can also promote their profibrotic phenotype. Kupffer cell derived CXCL16 recruits profibrotic NKT cells through its ligand receptor CXCR6 to the site of liver injury [
138].
In fibrogenesis and chronic liver injury, monocyte-derived macrophages display a similar functional switching from inflammatory/fibrogenic to pro-resolutive/antifibrogenic as described earlier in acute liver injury. Several observations from mouse models of hepatic fibrosis and its resolution support the phenotypic adaptation of macrophages. The selective depletion of early infiltrating Ly6C
hi macrophages reduces HSC activation and extracellular matrix (ECM) formation, while depletion of Ly6C
low macrophages during regression phases compromises ECM breakdown which preserves fibrosis [
67,
132]. Pharmacological blockade of Ly6C
hi infiltration using sophisticated Spiegelmer-based CCL-2 antagonists (artificial oligonucleotides specifically binding CCL-2), named mNOX-E36, augmented the proportion of pro-restorative Ly6C
low macrophages and accelerated fibrosis regression in animal models of chronic liver disease [
139]. Mechanistically, inflammatory Ly6C
hi monocyte-derived macrophages use TGF-β and IL-13 to activate HSCs [
67,
136,
140,
141,
142]. The phenotype switching from Ly6C
hi to Ly6C
low macrophages occurs after phagocytosis of dead cells (efferocytosis). The responsible signaling pathway comprises the receptor and tyrosine kinase Janus kinase (JAK) and signal transducer and activator of transcription (STAT) DNA-binding proteins. They mediate the signaling and downstream biological effects in response to binding of IL-10 and IL-6 [
69,
143,
144]. Additional macrophage receptors for efferocytosis and consequent phenotype switching are PtdSer-dependent receptor tyrosine kinases (RTKs) AXL and the proto-oncogene tyrosine-protein kinase MER (MERTK). Both are activated by IL-4 or IL-13 and lead to induction of anti-inflammatory and tissue repair responses in macrophages [
145].
5. Therapeutic Approaches
Numerous target points and approaches with promising results have been identified to clinically tackle liver disease initiation and their final common path of fibrosis and cirrhosis [
130,
147]. Macrophages represent sentinels of tissue homeostasis and immunological tolerance, are orchestrators in acute liver injury and hold dual, interchangeable functions in liver disease progression. Thus, targeting hepatic macrophages is one auspicious cornerstone of liver disease therapy. Generally, several basic approaches in targeting hepatic macrophages can be considered: (i) prohibit macrophage recruitment; (ii) inhibit macrophage activation; (iii) induce phenotype switching of macrophages. The transfusion of autologous macrophages in diseased patients (cell-based therapy) is another novel approach. In a very small, yet very innovative trial of autologous macrophage transfusion in chronic liver disease patients the safety and feasibility has been proven. An update on this new immunological branch of chronic liver disease has been nicely reviewed [
148,
149].
This entry is adapted from the peer-reviewed paper 10.3390/ijms22147249