The liver is an essential immunological organ due to its gatekeeper position to bypassing antigens from the intestinal blood flow and microbial products from the intestinal commensals. The tissue-resident liver macrophages, termed Kupffer cells, represent key phagocytes that closely interact with local parenchymal, interstitial and other immunological cells in the liver to maintain homeostasis and tolerance against harmless antigens. Upon liver injury, the pool of hepatic macrophages expands dramatically by infiltrating bone marrow-/monocyte-derived macrophages. The interplay of the injured microenvironment and altered macrophage pool skews the subsequent course of liver injuries. It may range from complete recovery to chronic inflammation, fibrosis, cirrhosis and eventually hepatocellular cancer.
- hepatic macrophages
- Kupffer cells
- monocyte-derived macrophages
- acute liver injury
- chronic liver injury
- NAFLD
- NASH
- liver fibrosis
- HBV
- HCV
1. Introduction
2. Hepatic Macrophages in Homeostasis
Under homeostatic conditions Kupffer cells, named after the German anatomist Karl Wilhelm von Kupffer (1829–1902) [12], are the predominate macrophage population in the liver. The initial notion that these non-migratory, tissue-resident, and self-sustaining phagocytes embryonically originate from hematopoietic stem cells [13] was recently challenged. Current data suggests that Kupffer cells derive from yolk sac erythromyeloid progenitors that colonize the fetal liver in embryonic day 8.5 in mice. At embryonic day 9.5 these cells give rise to macrophage precursors via signaling of the CX3C chemokine receptor 1 (CX3CR-1). Through upregulation of the transcription factor inhibitor of DNA binding 3 (ID3) the macrophage precursors eventually develop into Kupffer cells [14]. The half-life of a Kupffer cell is 12.4 days in mice, replenishment occurs through self-renewal which is tightly regulated by the transcription factors MafB and c-Maf [15]. Nonetheless, in case of Kupffer cell death or depletion, as it occurs in acute liver injury, circulating monocytes can contribute to the Kupffer cell pool as well [16] (Figure 1). This adaptability suggests that monocytes as hematopoietic derivates may develop into Kupffer Cells despite their distinct embryonic origin [17,18][17][18].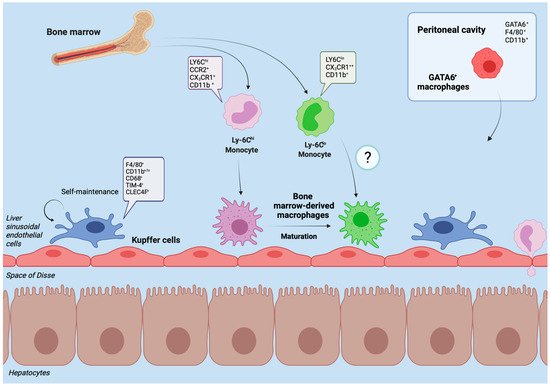
3. Hepatic Macrophages in Acute Liver Injury
Acute liver injury is defined clinically as a (more than) two- to threefold increase of liver transaminases above the upper limit of normal (marker of hepatocyte damage), jaundice and impaired coagulation function (International Normalized Ratio (INR) > 1.5) of hepatic origin in a patient without preexisting chronic liver disease. Acute liver failure additionally comprises altered mentation (termed hepatic encephalopathy) and is initiated by a severe acute liver injury [40][30], a life-threatening condition. The most common causes include hepatotoxic drugs (e.g., acetaminophen/paracetamol, phenprocoumon, antibiotics, antiepileptics), herbal or dietary supplements and acute viral hepatitis (hepatitis A (HAV), B (HBV) and E (HEV) viruses), although a multicenter data analysis from Germany revealed that in about one quarter of cases the cause remains unknown. Approximately one third to half of all patients with acute liver failure require emergency liver transplantation as a last resort [41,42][31][32]. The immense injury to the liver tissue greatly alters both, the microenvironment and immune cells. Danger-associated molecular patterns (DAMPs), a group of endogenous danger molecules released from damaged or dying cells such as high mobility group box 1 (HMGB1) or mitochondrial DNA (mtDNA) [43][33] are released and bind to pattern recognition receptors (PRR) such as TLRs on Kupffer cells, thereby activating them (Figure 2) [44][34]. The recognition of these danger signals leads to the formation of the inflammasome.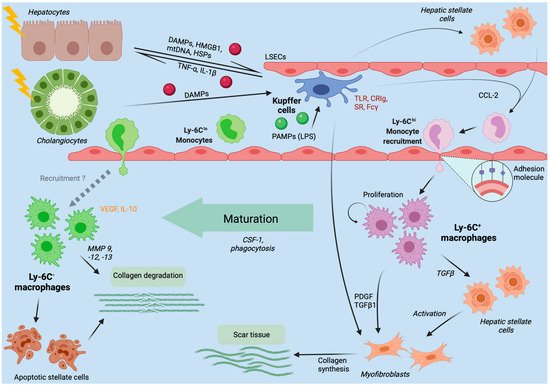
4. Chronic Liver Injury
4.1. Hepatic Macrophages in Nonalcoholic and Alcoholic Fatty Liver Disease
4.2. Hepatic Macrophages in Viral Hepatitis B and C
Chronic viral hepatitis caused by HBV or HCV remain a meaningful cause of liver associated morbidity and mortality [107][53]. The research on immunocompetent animal models is limited by the number of appropriate models as, for example, mice have a natural immunity against HCV and research on chimpanzees is hampered by financial and ethical constraints [108][54]. Similar to their multifaceted tasks in other liver diseases, hepatic macrophages can contribute to antiviral responses upon HBV or HCV infection. Research on the hepatotropic lymphocytic chroriomeningitis virus (LCMV) in mice has shown that the liver rapidly recruits pro-inflammatory macrophages (within 24 h) to support local Kupffer cells, when acutely infected [109][55]. In vitro cell culture studies of human Kupffer cells exposed to HBV surface antigen (HBsAg) have revealed increasing productions of the inflammatory cytokines TNF-α, IL-6 and CXCL8 (IL-8) that peaked after six hours of exposure [110][56]. Through NF-κB mediated transcription, these inflammatory cytokines, most importantly, IL-6 prohibit viral spreading in infected hepatocytes. IL-6 activates the mitogen-activated protein kinases exogenous signal-regulated kinase 1/2 (ERK1/2), and c-jun N-terminal kinase (JNK), two members of the mitogen-activated protein kinases (MAPKs) that transfer extracellular stimuli to a wide range of cellular stimuli [111][57]. ERK1/2 and JNK inhibit expression of hepatocyte nuclear factor (HNF) 1α and HNF4α, two transcription factors essential for HBV gene expression and replication [112][58]. Similarly, human Kupffer cells and monocyte-derived macrophages incubated with HCV in vitro activated the inflammasome and NF-κB via TLR2, which induced IL-1β and IL-18 secretion [113,114][59][60]. In line with the aforementioned two signal activation of inflammasomes, HCV exposed human macrophages showed: (i) viral RNA triggers MyD88-mediated TLR7 signaling to induce IL-1β mRNA expression; (ii) HCV uptake concomitantly induces a potassium efflux that activates the NLRP3 inflammasome for IL-1β processing and secretion; (iii) HCV infection is directly linked to liver inflammation by NLRP3 inflammasome activation [115][61]. Moreover, the depletion of Kupffer cells via clodronate-liposomes, a specific bisphosphonate that depletes macrophages [116][62], led to a rapid dissemination of LCMV in mice due to the inability to capture and process viral particles [117][63]. It is therefore appropriate to consider Kupffer cells a critical immune barrier in acute HBV and HCV infections.4.3. Fibrosis and Cirrhosis Modulation by Hepatic Macrophages
Fibrosis describes excessive scarring overproportionate to a wound healing response towards tissue injury [127][64]. If left untreated, progressive hepatic fibrosis leads to cirrhosis, a histoarchitectural remodeling with abundant collagen deposition that becomes clinically overt by decreasing hepatic function [128][65]. The chronic inflammation predisposes to carcinogenesis while increasing portal pressure triggers numerous clinical complications with significant morbidity and mortality [129][66]. Both, clinical observations and experimental models over the last years, challenged the former belief of irreversible liver fibrosis and identified new targets to halt and reverse this process [130][67]. Hepatic stellate cells (HSCs) transition to myofibroblasts and represent the main matrix-producing cells in the liver. Activation is induced by direct cell–cell interaction or binding of fibrogenic mediators [131][68]. In the dynamic process of inflammation and fibrogenesis, liver macrophages hold a dual function; during fibrogenesis the depletion of hepatic macrophages in mice improves scarring, whereas the depletion during resolution phases impedes adequate tissue restoration pointing towards functionally distinct subpopulations of macrophages within this process [132][69]. Upon damage to the hepatic microenvironment DAMPs and PAMPs are released that trigger local non-parenchymal cells (Kupffer cells, hepatic stellate cells, liver sinusoidal endothelial cells) to release a broad variety of inflammatory and profibrogenic soluble mediators. These mediators (e.g., CCL2) attract inflammatory immune cells like LyC6hi bone marrow-derived macrophages and activate matrix-producing profibrotic cell populations to form scar tissue [34,127][70][64]. Besides the recruitment of inflammatory and profibrotic macrophages by CCL2, Kupffer cells can also directly promote activation and survival of HSCs and myofibroblasts through secretion of growth factors (PDGF, TGF-β) and CCL5 [133,134,135,136,137][71][72][73][74][75]. The interaction of Kupffer cells with other immune cells, such as natural killer T (NKT) cells, can also promote their profibrotic phenotype. Kupffer cell derived CXCL16 recruits profibrotic NKT cells through its ligand receptor CXCR6 to the site of liver injury [138][76]. In fibrogenesis and chronic liver injury, monocyte-derived macrophages display a similar functional switching from inflammatory/fibrogenic to pro-resolutive/antifibrogenic as described earlier in acute liver injury. Several observations from mouse models of hepatic fibrosis and its resolution support the phenotypic adaptation of macrophages. The selective depletion of early infiltrating Ly6Chi macrophages reduces HSC activation and extracellular matrix (ECM) formation, while depletion of Ly6Clow macrophages during regression phases compromises ECM breakdown which preserves fibrosis [67,132][77][69]. Pharmacological blockade of Ly6Chi infiltration using sophisticated Spiegelmer-based CCL-2 antagonists (artificial oligonucleotides specifically binding CCL-2), named mNOX-E36, augmented the proportion of pro-restorative Ly6Clow macrophages and accelerated fibrosis regression in animal models of chronic liver disease [139][78]. Mechanistically, inflammatory Ly6Chi monocyte-derived macrophages use TGF-β and IL-13 to activate HSCs [67,136,140,141,142][77][74][79][80][81]. The phenotype switching from Ly6Chi to Ly6Clow macrophages occurs after phagocytosis of dead cells (efferocytosis). The responsible signaling pathway comprises the receptor and tyrosine kinase Janus kinase (JAK) and signal transducer and activator of transcription (STAT) DNA-binding proteins. They mediate the signaling and downstream biological effects in response to binding of IL-10 and IL-6 [69,143,144][82][83][84]. Additional macrophage receptors for efferocytosis and consequent phenotype switching are PtdSer-dependent receptor tyrosine kinases (RTKs) AXL and the proto-oncogene tyrosine-protein kinase MER (MERTK). Both are activated by IL-4 or IL-13 and lead to induction of anti-inflammatory and tissue repair responses in macrophages [145][85].5. Therapeutic Approaches
Numerous target points and approaches with promising results have been identified to clinically tackle liver disease initiation and their final common path of fibrosis and cirrhosis [130,147][67][86]. Macrophages represent sentinels of tissue homeostasis and immunological tolerance, are orchestrators in acute liver injury and hold dual, interchangeable functions in liver disease progression. Thus, targeting hepatic macrophages is one auspicious cornerstone of liver disease therapy. Generally, several basic approaches in targeting hepatic macrophages can be considered: (i) prohibit macrophage recruitment; (ii) inhibit macrophage activation; (iii) induce phenotype switching of macrophages. The transfusion of autologous macrophages in diseased patients (cell-based therapy) is another novel approach. In a very small, yet very innovative trial of autologous macrophage transfusion in chronic liver disease patients the safety and feasibility has been proven. An update on this new immunological branch of chronic liver disease has been nicely reviewed [148,149][87][88].References
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321.
- Bilzer, M.; Roggel, F.; Gerbes, A.L. Role of Kupffer cells in host defense and liver disease. Liver Int. 2006, 26, 1175–1186.
- Guilliams, M.; Dutertre, C.A.; Scott, C.L.; McGovern, N.; Sichien, D.; Chakarov, S.; Van Gassen, S.; Chen, J.; Poidinger, M.; De Prijck, S.; et al. Unsupervised High-Dimensional Analysis Aligns Dendritic Cells across Tissues and Species. Immunity 2016, 45, 669–684.
- Wen, Y.; Lambrecht, J.; Ju, C.; Tacke, F. Hepatic macrophages in liver homeostasis and diseases-diversity, plasticity and therapeutic opportunities. Cell. Mol. Immunol. 2021, 18, 45–56.
- Abdullah, Z.; Knolle, P.A. Liver macrophages in healthy and diseased liver. Pflugers Arch. 2017, 469, 553–560.
- Gola, A.; Dorrington, M.G.; Speranza, E.; Sala, C.; Shih, R.M.; Radtke, A.J.; Wong, H.S.; Baptista, A.P.; Hernandez, J.M.; Castellani, G.; et al. Commensal-driven immune zonation of the liver promotes host defence. Nature 2021, 589, 131–136.
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566.
- Tacke, F. Targeting hepatic macrophages to treat liver diseases. J. Hepatol. 2017, 66, 1300–1312.
- Heymann, F.; Peusquens, J.; Ludwig-Portugall, I.; Kohlhepp, M.; Ergen, C.; Niemietz, P.; Martin, C.; van Rooijen, N.; Ochando, J.C.; Randolph, G.J.; et al. Liver inflammation abrogates immunological tolerance induced by Kupffer cells. Hepatology 2015, 62, 279–291.
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321.
- Zigmond, E.; Samia-Grinberg, S.; Pasmanik-Chor, M.; Brazowski, E.; Shibolet, O.; Halpern, Z. Infiltrating monocyte-derived macrophages and resident kupffer cells display different ontogeny and functions in acute liver injury. J. Immunol. 2014, 193, 344–353.
- von Kupffer, K. Über Sternzellen der Leber. Arch. Mikroskop Anat. 1876, 12, 353–358.
- van Furth, R.; Cohn, Z.A. The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 1968, 128, 415–435.
- Horst, A.K.; Tiegs, G.; Diehl, L. Contribution of Macrophage Efferocytosis to Liver Homeostasis and Disease. Front. Immunol. 2019, 10, 2670.
- Soucie, E.L.; Weng, Z.; Geirsdottir, L.; Molawi, K.; Maurizio, J.; Fenouil, R.; Mossadegh-Keller, N.; Gimenez, G.; VanHille, L.; Beniazza, M.; et al. Lineage-specific enhancers activate self-renewal genes in macrophages and embryonic stem cells. Science 2016, 351, aad5510.
- Bonnardel, J.; T’Jonck, W.; Gaublomme, D.; Browaeys, R.; Scott, C.L.; Martens, L.; Vanneste, B.; De Prijck, S.; Nedospasov, S.A.; Kremer, A.; et al. Stellate Cells, Hepatocytes, and Endothelial Cells Imprint the Kupffer Cell Identity on Monocytes Colonizing the Liver Macrophage Niche. Immunity 2019, 51, 638–654.e9.
- Guilliams, M.; Scott, C.L. Does niche competition determine the origin of tissue-resident macrophages? Nat. Rev. Immunol. 2017, 17, 451–460.
- David, B.A.; Rezende, R.M.; Antunes, M.M.; Santos, M.M.; Freitas Lopes, M.A.; Diniz, A.B.; Sousa Pereira, R.V.; Marchesi, S.C.; Alvarenga, D.M.; Nakagaki, B.N.; et al. Combination of Mass Cytometry and Imaging Analysis Reveals Origin, Location, and Functional Repopulation of Liver Myeloid Cells in Mice. Gastroenterology 2016, 151, 1176–1191.
- Varol, C.; Mildner, A.; Jung, S. Macrophages: Development and tissue specialization. Annu. Rev. Immunol. 2015, 33, 643–675.
- Strnad, P.; Tacke, F.; Koch, A.; Trautwein, C. Liver—Guardian, modifier and target of sepsis. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 55–66.
- Terpstra, V.; van Berkel, T.J. Scavenger receptors on liver Kupffer cells mediate the in vivo uptake of oxidatively damaged red blood cells in mice. Blood 2000, 95, 2157–2163.
- Theurl, I.; Hilgendorf, I.; Nairz, M.; Tymoszuk, P.; Haschka, D.; Asshoff, M.; He, S.; Gerhardt, L.M.; Holderried, T.A.; Seifert, M.; et al. On-demand erythrocyte disposal and iron recycling requires transient macrophages in the liver. Nat. Med. 2016, 22, 945–951.
- Scott, C.L.; Guilliams, M. The role of Kupffer cells in hepatic iron and lipid metabolism. J. Hepatol. 2018, 69, 1197–1199.
- Kanamori, Y.; Tanaka, M.; Itoh, M.; Ochi, K.; Ito, A.; Hidaka, I.; Sakaida, I.; Ogawa, Y.; Suganami, T. Iron-rich Kupffer cells exhibit phenotypic changes during the development of liver fibrosis in NASH. iScience 2021, 24, 102032.
- van Vliet, S.J.; Gringhuis, S.I.; Geijtenbeek, T.B.H.; van Kooyk, Y. Regulation of effector T cells by antigen-presenting cells via interaction of the C-type lectin MGL with CD45. Nat. Immunol. 2006, 7, 1200–1208.
- Deppermann, C.; Kratofil, R.M.; Peiseler, M.; David, B.A.; Zindel, J.; Castanheira, F.; van der Wal, F.; Carestia, A.; Jenne, C.N.; Marth, J.D.; et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 2020, 217, e20190723.
- Wang, Y.; van der Tuin, S.; Tjeerdema, N.; van Dam, A.D.; Rensen, S.S.; Hendrikx, T.; Berbée, J.F.; Atanasovska, B.; Fu, J.; Hoekstra, M.; et al. Plasma cholesteryl ester transfer protein is predominantly derived from Kupffer cells. Hepatology 2015, 62, 1710–1722.
- Thompson, A.; Di Angelantonio, E.; Sarwar, N.; Erqou, S.; Saleheen, D.; Dullaart, R.P.; Keavney, B.; Ye, Z.; Danesh, J. Association of cholesteryl ester transfer protein genotypes with CETP mass and activity, lipid levels, and coronary risk. JAMA 2008, 299, 2777–2788.
- van der Tuin, S.J.L.; Li, Z.; Berbee, J.F.P.; Verkouter, I.; Ringnalda, L.E.; Neele, A.E.; van Klinken, J.B.; Rensen, S.S.; Fu, J.; de Winther, M.P.J.; et al. Lipopolysaccharide Lowers Cholesteryl Ester Transfer Protein by Activating F4/80(+)Clec4f(+)Vsig4(+)Ly6C(-) Kupffer Cell Subsets. J. Am. Heart Assoc. 2018, 7, e008105.
- European Association for the Study of the Liver; Wendon, J.; Panel, M.; Cordoba, J.; Dhawan, A.; Larsen, F.S.; Manns, M.; Samuel, D.; Simpson, K.J. EASL Clinical Practical Guidelines on the management of acute (fulminant) liver failure. J. Hepatol. 2017, 66, 1047–1081.
- Bernal, W.; Wendon, J. Acute Liver Failure. N. Engl. J. Med. 2013, 369, 2525–2534.
- Hadem, J.; Tacke, F.; Bruns, T.; Langgartner, J.; Strnad, P.; Denk, G.U.; Fikatas, P.; Manns, M.P.; Hofmann, W.P.; Gerken, G.; et al. Etiologies and outcomes of acute liver failure in Germany. Clin. Gastroenterol. Hepatol. 2012, 10, 664–669.e2.
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immun. Netw. 2018, 18, e27.
- Mihm, S. Danger-Associated Molecular Patterns (DAMPs): Molecular Triggers for Sterile Inflammation in the Liver. Int. J. Mol. Sci. 2018, 19, 3104.
- Weiskirchen, R.; Tacke, F. Liver Fibrosis: From Pathogenesis to Novel Therapies. Dig. Dis. 2016, 34, 410–422.
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Karlsen, T.H.; Tacke, F. The times they are a’changin—Positioning the European Association for the Study of the Liver in the changing landscape of hepatology. J. Hepatol. 2018, 68, 873–875.
- Estes, C.; Razavi, H.; Loomba, R.; Younossi, Z.; Sanyal, A.J. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology 2018, 67, 123–133.
- Di Cesare, M.; Sorić, M.; Bovet, P.; Miranda, J.J.; Bhutta, Z.; Stevens, G.A.; Laxmaiah, A.; Kengne, A.-P.; Bentham, J. The epidemiological burden of obesity in childhood: A worldwide epidemic requiring urgent action. BMC Med. 2019, 17, 212.
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402.
- Taylor, R.S.; Taylor, R.J.; Bayliss, S.; Hagström, H.; Nasr, P.; Schattenberg, J.M.; Ishigami, M.; Toyoda, H.; Wai-Sun Wong, V.; Peleg, N.; et al. Association Between Fibrosis Stage and Outcomes of Patients with Nonalcoholic Fatty Liver Disease: A Systematic Review and Meta-Analysis. Gastroenterology 2020, 158, 1611–1625.e12.
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565.
- Povsic, M.; Wong, O.Y.; Perry, R.; Bottomley, J. A Structured Literature Review of the Epidemiology and Disease Burden of Non-Alcoholic Steatohepatitis (NASH). Adv. Ther. 2019, 36, 1574–1594.
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wai-Sun Wong, V.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Alvarez-Quiñones Sanz, M.; Conde-Martin, A.F.; et al. Fibrosis Severity as a Determinant of Cause-Specific Mortality in Patients With Advanced Nonalcoholic Fatty Liver Disease: A Multi-National Cohort Study. Gastroenterology 2018, 155, 443–457.e17.
- Byrne, C.D.; Targher, G. NAFLD: A multisystem disease. J. Hepatol. 2015, 62, S47–S64.
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965.
- Knorr, J.; Wree, A.; Tacke, F.; Feldstein, A.E. The NLRP3 Inflammasome in Alcoholic and Nonalcoholic Steatohepatitis. Semin. Liver. Dis. 2020, 40, 298–306.
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-Induced Signaling Causes Release of Inflammatory Extracellular Vesicles From Hepatocytes. Gastroenterology 2016, 150, 956–967.
- Bartneck, M.; Fech, V.; Ehling, J.; Govaere, O.; Warzecha, K.T.; Hittatiya, K.; Vucur, M.; Gautheron, J.; Luedde, T.; Trautwein, C.; et al. Histidine-rich glycoprotein promotes macrophage activation and inflammation in chronic liver disease. Hepatology 2016, 63, 1310–1324.
- Rolny, C.; Mazzone, M.; Tugues, S.; Laoui, D.; Johansson, I.; Coulon, C.; Squadrito, M.L.; Segura, I.; Li, X.; Knevels, E.; et al. HRG Inhibits Tumor Growth and Metastasis by Inducing Macrophage Polarization and Vessel Normalization through Downregulation of PlGF. Cancer Cell 2011, 19, 31–44.
- Bereshchenko, O.; Migliorati, G.; Bruscoli, S.; Riccardi, C. Glucocorticoid-Induced Leucine Zipper: A Novel Anti-inflammatory Molecule. Front. Pharmacol. 2019, 10, 308.
- Robert, O.; Boujedidi, H.; Bigorgne, A.; Ferrere, G.; Voican, C.S.; Vettorazzi, S.; Tuckermann, J.P.; Bouchet-Delbos, L.; Tran, T.; Hemon, P.; et al. Decreased expression of the glucocorticoid receptor-GILZ pathway in Kupffer cells promotes liver inflammation in obese mice. J. Hepatol. 2016, 64, 916–924.
- Wiktor, S.Z.; Hutin, Y.J. The global burden of viral hepatitis: Better estimates to guide hepatitis elimination efforts. Lancet 2016, 388, 1030–1031.
- Vercauteren, K.; de Jong, Y.P.; Meuleman, P. HCV animal models and liver disease. J. Hepatol. 2014, 61, S26–S33.
- Movita, D.; van de Garde, M.D.; Biesta, P.; Kreefft, K.; Haagmans, B.; Zuniga, E.; Herschke, F.; De Jonghe, S.; Janssen, H.L.; Gama, L.; et al. Inflammatory monocytes recruited to the liver within 24 h after virus-induced inflammation resemble Kupffer cells but are functionally distinct. J. Virol. 2015, 89, 4809–4817.
- Boltjes, A.; van Montfoort, N.; Biesta, P.J.; Op den Brouw, M.L.; Kwekkeboom, J.; van der Laan, L.J.W.; Janssen, H.L.A.; Boonstra, A.; Woltman, A.M. Kupffer Cells Interact With Hepatitis B Surface Antigen In Vivo and In Vitro, Leading to Proinflammatory Cytokine Production and Natural Killer Cell Function. J. Infect. Dis. 2014, 211, 1268–1278.
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83.
- Hösel, M.; Quasdorff, M.; Wiegmann, K.; Webb, D.; Zedler, U.; Broxtermann, M.; Tedjokusumo, R.; Esser, K.; Arzberger, S.; Kirschning, C.J.; et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology 2009, 50, 1773–1782.
- Shrivastava, S.; Mukherjee, A.; Ray, R.; Ray, R.B. Hepatitis C virus induces interleukin-1β (IL-1β)/IL-18 in circulatory and resident liver macrophages. J. Virol. 2013, 87, 12284–12290.
- Chang, S.; Dolganiuc, A.; Szabo, G. Toll-like receptors 1 and 6 are involved in TLR2-mediated macrophage activation by hepatitis C virus core and NS3 proteins. J. Leukoc. Biol. 2007, 82, 479–487.
- Negash, A.A.; Ramos, H.J.; Crochet, N.; Lau, D.T.; Doehle, B.; Papic, N.; Delker, D.A.; Jo, J.; Bertoletti, A.; Hagedorn, C.H.; et al. IL-1β production through the NLRP3 inflammasome by hepatic macrophages links hepatitis C virus infection with liver inflammation and disease. PLoS Pathog. 2013, 9, e1003330.
- Frith, J.C.; Mönkkönen, J.; Blackburn, G.M.; Russell, R.G.G.; Rogers, M.J. Clodronate and Liposome-Encapsulated Clodronate Are Metabolized to a Toxic ATP Analog, Adenosine 5′-(β,γ-Dichloromethylene) Triphosphate, by Mammalian Cells In Vitro. J. Bone Miner. Res. 1997, 12, 1358–1367.
- Lang, P.A.; Recher, M.; Honke, N.; Scheu, S.; Borkens, S.; Gailus, N.; Krings, C.; Meryk, A.; Kulawik, A.; Cervantes-Barragan, L.; et al. Tissue macrophages suppress viral replication and prevent severe immunopathology in an interferon-I-dependent manner in mice. Hepatology 2010, 52, 25–32.
- Weiskirchen, R.; Weiskirchen, S.; Tacke, F. Organ and tissue fibrosis: Molecular signals, cellular mechanisms and translational implications. Mol. Aspects Med. 2019, 65, 2–15.
- Maurice, J.; Pinzani, M. The stratification of cirrhosis. Hepatol. Res. 2020, 50, 535–541.
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J. Hepatol. 2018, 69, 406–460.
- Tacke, F.; Weiskirchen, R. Non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH)-related liver fibrosis: Mechanisms, treatment and prevention. Ann. Transl. Med. 2021, 9, 729.
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411.
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65.
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110.
- Liu, W.; Baker, R.D.; Bhatia, T.; Zhu, L.; Baker, S.S. Pathogenesis of nonalcoholic steatohepatitis. Cell Mol. Life Sci. 2016, 73, 1969–1987.
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2020, 31, 406–421.e7.
- Kocabayoglu, P.; Lade, A.; Lee, Y.A.; Dragomir, A.C.; Sun, X.; Fiel, M.I.; Thung, S.; Aloman, C.; Soriano, P.; Hoshida, Y.; et al. β-PDGF receptor expressed by hepatic stellate cells regulates fibrosis in murine liver injury, but not carcinogenesis. J. Hepatol. 2015, 63, 141–147.
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473.
- Sasaki, R.; Devhare, P.B.; Steele, R.; Ray, R.; Ray, R.B. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757.
- Wehr, A.; Baeck, C.; Heymann, F.; Niemietz, P.M.; Hammerich, L.; Martin, C.; Zimmermann, H.W.; Pack, O.; Gassler, N.; Hittatiya, K.; et al. Chemokine receptor CXCR6-dependent hepatic NK T Cell accumulation promotes inflammation and liver fibrosis. J. Immunol. 2013, 190, 5226–5236.
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195.
- Baeck, C.; Wei, X.; Bartneck, M.; Fech, V.; Heymann, F.; Gassler, N.; Hittatiya, K.; Eulberg, D.; Luedde, T.; Trautwein, C.; et al. Pharmacological inhibition of the chemokine C-C motif chemokine ligand 2 (monocyte chemoattractant protein 1) accelerates liver fibrosis regression by suppressing Ly-6C(+) macrophage infiltration in mice. Hepatology 2014, 59, 1060–1072.
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274.
- Liu, Y.; Munker, S.; Müllenbach, R.; Weng, H.-L. IL-13 Signaling in Liver Fibrogenesis. Front. Immunol. 2012, 3, 116.
- Liaskou, E.; Zimmermann, H.W.; Li, K.K.; Oo, Y.H.; Suresh, S.; Stamataki, Z.; Qureshi, O.; Lalor, P.F.; Shaw, J.; Syn, W.K.; et al. Monocyte subsets in human liver disease show distinct phenotypic and functional characteristics. Hepatology 2013, 57, 385–398.
- Wang, M.; You, Q.; Lor, K.; Chen, F.; Gao, B.; Ju, C. Chronic alcohol ingestion modulates hepatic macrophage populations and functions in mice. J. Leukoc. Biol. 2014, 96, 657–665.
- Salas, A.; Hernandez-Rocha, C.; Duijvestein, M.; Faubion, W.; McGovern, D.; Vermeire, S. JAK–STAT pathway targeting for the treatment of inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 323–337.
- Campana, L.; Starkey Lewis, P.J.; Pellicoro, A.; Aucott, R.L.; Man, J.; O’Duibhir, E.; Mok, S.E.; Ferreira-Gonzalez, S.; Livingstone, E.; Greenhalgh, S.N.; et al. The STAT3–IL-10–IL-6 Pathway Is a Novel Regulator of Macrophage Efferocytosis and Phenotypic Conversion in Sterile Liver Injury. J. Immunol. 2018, 200, 1169–1187.
- Bosurgi, L.; Cao, Y.G.; Cabeza-Cabrerizo, M.; Tucci, A.; Hughes, L.D.; Kong, Y.; Weinstein, J.S.; Licona-Limon, P.; Schmid, E.T.; Pelorosso, F.; et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 2017, 356, 1072–1076.
- Lambrecht, J.; van Grunsven, L.A.; Tacke, F. Current and emerging pharmacotherapeutic interventions for the treatment of liver fibrosis. Expert Opin. Pharmacother. 2020, 21, 1637–1650.
- Moroni, F.; Dwyer, B.J.; Graham, C.; Pass, C.; Bailey, L.; Ritchie, L.; Mitchell, D.; Glover, A.; Laurie, A.; Doig, S.; et al. Safety profile of autologous macrophage therapy for liver cirrhosis. Nat. Med. 2019, 25, 1560–1565.
- Starkey Lewis, P.J.; Moroni, F.; Forbes, S.J. Macrophages as a Cell-Based Therapy for Liver Disease. Semin. Liver. Dis. 2019, 39, 442–451.