 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Ashraf Yusuf Rangrez | + 2050 word(s) | 2050 | 2021-07-09 06:25:05 | | | |
| 2 | Lindsay Dong | Meta information modification | 2050 | 2021-07-27 04:30:12 | | |
Video Upload Options
One of the key proteins involved in stress-mediated cardiomyocyte signal transduction is a small GTPase RhoA. Importantly, the regulation of RhoA activation is critical for effective immune cell response and is being considered as one of the potential therapeutic targets in many immune-cell-mediated inflammatory diseases.
1. Introduction
The immune system is based on two main complexes: (1) the innate immune system and (2) the adaptive immune system. The innate immune system is triggered by direct contact with pathogens or inflammatory/danger signals and includes non-cellular responses, i.e., the release of inflammatory cytokines, and cellular responses, i.e., infiltration of innate immune cells (macrophages, dendritic cells, and granulocytes) into affected tissue [1]. The adaptive immune system also involves non-cellular (hormonal) and cellular responses (stimulation of B- and T-cells), but in contrast to the innate response, it builds up an “immunologic memory” by developing pathogen-specific receptors [1]. RhoA (ras homolog family member A), an ubiquitously expressed small GTPase, acts as a molecular switch not only in the activation of cytoskeletal proteins but also in responding chemokines, cytokines, and growth factors released from both innate and adaptive immune cells [2]. Furthermore, RhoA activation and RhoA-dependent signaling pathways in cardiomyocytes [3] and in immune cells [2][4] have been shown to mediate immune responses, which play an important role in pathogenesis and progression of cardiac dysfunction [1].
2. Links between Cardiac Hypertrophy, Heart Failure, and Immune Cell Activation
In the last decades, the role of the innate and adaptive immune response is being linked with a number of signaling molecules and pathways, including RhoA activation in cardiomyocytes [2][5]. Furthermore, it has been illustrated that different cardiac diseases (e.g., ischemic, hypertensive, and genetic cardiomyopathies) converge in inducing a common immune response that contributes to disease progression [1].
Signals that trigger activation of innate immune cells and subsequent immune responses are pathogen-associated molecules or “danger-molecules” and other signals arising from damaged tissue [1]. The main group of receptors for inflammatory signals consists of pattern recognition receptors (PRRs) [1]. PRRs, which are generally upregulated in HF [6], are commonly expressed by immune cells, but have also been found in cardiac cells [7][8]. Although PPRs are best known for their activation after contact with pathogens, it can also be activated by danger-associated molecular patterns (DAMPs) that are released by damaged or dying cells [9], e.g., injured myocardium. The activation of PPRs triggers the release of inflammatory cytokines [10] (e.g., interferones (IFN) and IL-1β) and thus accelerates immune responses [1] (Figure 1).
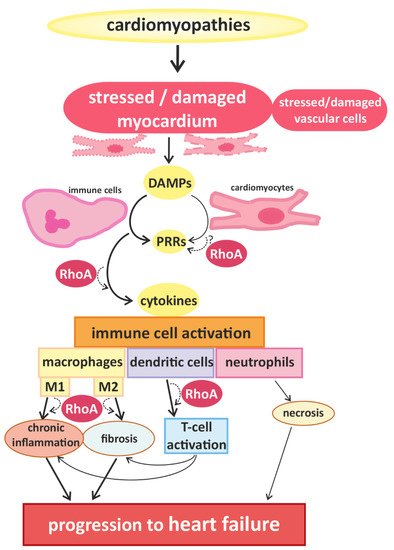
3. RhoA Activation and Signaling in Immune Cells
Recent studies demonstrate that RhoA plays an important role in immune responses, with its effects being highly dependent on the spatio-temporal regulation of RhoA activation in different innate and adaptive immune cells [2][27]. On molecular level, the activation of RhoA in immune cells and cardiomyocytes depends on its change from a guanosine diphosphate (GDP)-bound “inactive” to a guanosine triphosphate (GTP)-bound “active” state and back [28]. This cycle is regulated by regulatory proteins, namely guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), guanine nucleotide dissociation inhibitors (GDIs) and GDI dissociation factors (GDFs) [5]. GEFs facilitate the dissociation of GDP from RhoA, thus accelerating the binding of GTP and allowing the activation of downstream effectors [29]. GAPs catalyze the hydrolysis of GTP back to GDP and thus the release of the effector [30]. GDIs disconnect RhoA from the plasma membrane, thus inhibiting the dissociation of GDP [31], while GDFs initiate the dissociation of GDIs from RhoA allowing the cycle to start again [5]. A number of these regulators have been shown to be identical in immune cells and cardiomyocytes (e.g., LARG (leukemia-associated RhoGEF), GEF-H1/Lfc (Lbc’s first cousin), PDZ-RhoGEF, p190RhoGAP, and Vav1) [32][33], while others were found specifically in immune cells (e.g., RhoA-GAP, Myo9B, and Rho-GEF7) [34]. Even some of the up- and downstream signaling pathways of RhoA described in cardiomyocytes have been proven to play an important role in immune cells, e.g., RhoA activation via Gα12/13-coupled membrane receptors [35][36][37] and RhoGEF or activation of the RhoA effector, ROCK [2][4][35][36][37][38].
RhoA activation has also been associated with activation of β2-adrenergic receptors (β2-AR), probably via p115RhoGEF [39][40]. Hyperactivity of the sympathetic nervous system is one of the hallmarks of heart failure that involves catecholamine spillover, and is associated with pro-inflammatory signaling [41][42][43]. Several studies have shown that infusion of isoprenaline—a synthetic catecholamine and β2-AR agonist—in mice induces cardiac inflammation and dysfunction [44], and infusion of noradrenalin, also a β2-AR agonist, induces cardiac hypertrophy and fibrosis in rats [45]. However, β1-ARs have been shown to be ubiquitously expressed in rodent (ventricular) cardiomyocytes, while β2-ARs were only found in a very small percentage of these myocytes [46]. In contrast, β2-ARs were found to be abundant in non-myocytes of rodent heart tissue [46], suggesting that endothelial cells, fibroblasts and/or immune cells in cardiac tissue likely express β2-ARs.
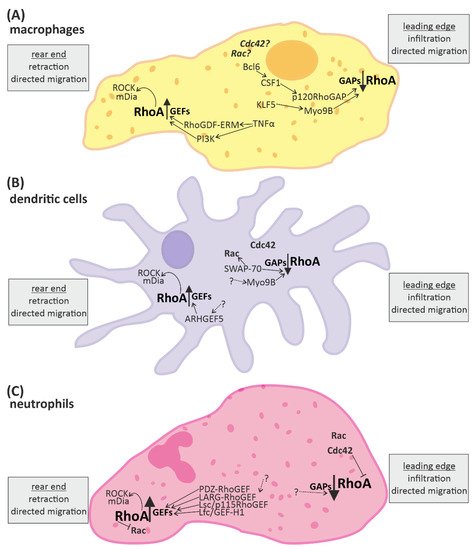
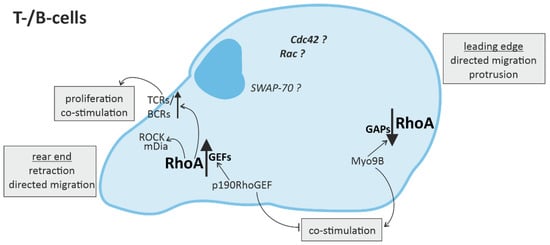
4. Conclusions
References
- Frantz, S.; Falcao-Pires, I.; Balligand, J.L.; Bauersachs, J.; Brutsaert, D.; Ciccarelli, M.; Dawson, D.; de Windt, L.J.; Giacca, M.; Hamdani, N.; et al. The innate immune system in chronic cardiomyopathy: A European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur. J. Heart Fail. 2018, 20, 445–459.
- Bros, M.; Haas, K.; Moll, L.; Grabbe, S. RhoA as a Key Regulator of Innate and Adaptive Immunity. Cells 2019, 8, 733.
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407.
- Bodin, S.; Welch, M.D. Plasma membrane organization is essential for balancing competing pseudopod- and uropod-promoting signals during neutrophil polarization and migration. Mol. Biol. Cell 2005, 16, 5773–5783.
- Kilian, L.S.; Voran, J.; Frank, D.; Rangrez, A.Y. RhoA: A dubious molecule in cardiac pathophysiology. J. Biomed. Sci. 2021, 28, 33.
- Frantz, S.; Nahrendorf, M. Cardiac macrophages and their role in ischaemic heart disease. Cardiovasc. Res. 2014, 102, 240–248.
- Nishimura, M.; Naito, S. Tissue-specific mRNA expression profiles of human toll-like receptors and related genes. Biol. Pharm. Bull. 2005, 28, 886–892.
- Yin, Y.; Yan, Y.; Jiang, X.; Mai, J.; Chen, N.C.; Wang, H.; Yang, X.F. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int. J. Immunopathol. Pharmacol. 2009, 22, 311–322.
- Lin, L.; Knowlton, A.A. Innate immunity and cardiomyocytes in ischemic heart disease. Life Sci. 2014, 100, 1–8.
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650.
- Rangrez, A.Y.; Eden, M.; Poyanmehr, R.; Kuhn, C.; Stiebeling, K.; Dierck, F.; Bernt, A.; Lullmann-Rauch, R.; Weiler, H.; Kirchof, P.; et al. Myozap Deficiency Promotes Adverse Cardiac Remodeling via Differential Regulation of Mitogen-activated Protein Kinase/Serum-response Factor and beta-Catenin/GSK-3beta Protein Signaling. J. Biol. Chem. 2016, 291, 4128–4143.
- De Vito, P. Atrial natriuretic peptide: An old hormone or a new cytokine? Peptides 2014, 58, 108–116.
- Hulsmans, M.; Sam, F.; Nahrendorf, M. Monocyte and macrophage contributions to cardiac remodeling. J. Mol. Cell. Cardiol. 2016, 93, 149–155.
- Sansonetti, M.; Waleczek, F.J.G.; Jung, M.; Thum, T.; Perbellini, F. Resident cardiac macrophages: Crucial modulators of cardiac (patho)physiology. Basic Res. Cardiol. 2020, 115, 77.
- Liu, Y.; Minze, L.J.; Mumma, L.; Li, X.C.; Ghobrial, R.M.; Kloc, M. Mouse macrophage polarity and ROCK1 activity depend on RhoA and non-apoptotic Caspase 3. Exp. Cell Res. 2016, 341, 225–236.
- Hazebroek, M.R.; Moors, S.; Dennert, R.; van den Wijngaard, A.; Krapels, I.; Hoos, M.; Verdonschot, J.; Merken, J.J.; de Vries, B.; Wolffs, P.F.; et al. Prognostic Relevance of Gene-Environment Interactions in Patients with Dilated Cardiomyopathy: Applying the MOGE(S) Classification. J. Am. Coll. Cardiol. 2015, 66, 1313–1323.
- Lynch, T.L.t.; Ismahil, M.A.; Jegga, A.G.; Zilliox, M.J.; Troidl, C.; Prabhu, S.D.; Sadayappan, S. Cardiac inflammation in genetic dilated cardiomyopathy caused by MYBPC3 mutation. J. Mol. Cell. Cardiol. 2017, 102, 83–93.
- Felix, S.B.; Beug, D.; Dorr, M. Immunoadsorption therapy in dilated cardiomyopathy. Expert Rev. Cardiovasc. Ther. 2015, 13, 145–152.
- Mortensen, R.M. Immune cell modulation of cardiac remodeling. Circulation 2012, 125, 1597–1600.
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245.
- Laroumanie, F.; Douin-Echinard, V.; Pozzo, J.; Lairez, O.; Tortosa, F.; Vinel, C.; Delage, C.; Calise, D.; Dutaur, M.; Parini, A.; et al. CD4+ T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 2014, 129, 2111–2124.
- Nevers, T.; Salvador, A.M.; Grodecki-Pena, A.; Knapp, A.; Velazquez, F.; Aronovitz, M.; Kapur, N.K.; Karas, R.H.; Blanton, R.M.; Alcaide, P. Left Ventricular T-Cell Recruitment Contributes to the Pathogenesis of Heart Failure. Circulation Heart Fail. 2015, 8, 776–787.
- Ogawa, T.; Veinot, J.P.; Davies, R.A.; Haddad, H.; Smith, S.J.; Masters, R.G.; Hendry, P.J.; Starling, R.; de Bold, M.K.; Ponce, A.; et al. Neuroendocrine profiling of humans receiving cardiac allografts. J. Heart Lung Transplant. 2005, 24, 1046–1054.
- Kiemer, A.K.; Weber, N.C.; Furst, R.; Bildner, N.; Kulhanek-Heinze, S.; Vollmar, A.M. Inhibition of p38 MAPK activation via induction of MKP-1: Atrial natriuretic peptide reduces TNF-alpha-induced actin polymerization and endothelial permeability. Circ. Res. 2002, 90, 874–881.
- Mtairag el, M.; Houard, X.; Rais, S.; Pasquier, C.; Oudghiri, M.; Jacob, M.P.; Meilhac, O.; Michel, J.B. Pharmacological potentiation of natriuretic peptide limits polymorphonuclear neutrophil-vascular cell interactions. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1824–1831.
- Chen, W.; Spitzl, A.; Mathes, D.; Nikolaev, V.O.; Werner, F.; Weirather, J.; Spiranec, K.; Rock, K.; Fischer, J.W.; Kammerer, U.; et al. Endothelial Actions of ANP Enhance Myocardial Inflammatory Infiltration in the Early Phase After Acute Infarction. Circ. Res. 2016, 119, 237–248.
- Hind, L.E.; Vincent, W.J.B.; Huttenlocher, A. Leading from the Back: The Role of the Uropod in Neutrophil Polarization and Migration. Dev. Cell 2016, 38, 161–169.
- Jaffe, A.B.; Hall, A. Rho GTPases: Biochemistry and biology. Annu. Rev. Cell. Dev. Biol. 2005, 21, 247–269.
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877.
- Verma, S.K.; Krishnamurthy, P.; Barefield, D.; Singh, N.; Gupta, R.; Lambers, E.; Thal, M.; Mackie, A.; Hoxha, E.; Ramirez, V.; et al. Interleukin-10 treatment attenuates pressure overload-induced hypertrophic remodeling and improves heart function via signal transducers and activators of transcription 3-dependent inhibition of nuclear factor-kappaB. Circulation 2012, 126, 418–429.
- Fukumoto, Y.; Kaibuchi, K.; Hori, Y.; Fujioka, H.; Araki, S.; Ueda, T.; Kikuchi, A.; Takai, Y. Molecular cloning and characterization of a novel type of regulatory protein (GDI) for the rho proteins, ras p21-like small GTP-binding proteins. Oncogene 1990, 5, 1321–1328.
- Fine, N.; Dimitriou, I.D.; Rottapel, R. Go with the flow: GEF-H1 mediated shear stress mechanotransduction in neutrophils. Small GTPases 2020, 11, 23–31.
- Shi, Y.Q.; Zhang, J.Y.; Mullin, M.; Dong, B.X.; Alberts, A.S.; Siminovitch, K.A. The mDial formin is required for neutrophil polarization, migration, and activation of the LARG/RhoA/ROCK signaling axis during chemotaxis. J. Immunol. 2009, 182, 3837–3845.
- Chen, W.; Ghobrial, R.M.; Li, X.C.; Kloc, M. Inhibition of RhoA and mTORC2/Rictor by Fingolimod (FTY720) induces p21-activated kinase 1, PAK-1 and amplifies podosomes in mouse peritoneal macrophages. Immunobiology 2018, 223, 634–647.
- Francis, S.A.; Shen, X.; Young, J.B.; Kaul, P.; Lerner, D.J. Rho GEF Lsc is required for normal polarization, migration, and adhesion of formyl-peptide-stimulated neutrophils. Blood 2006, 107, 1627–1635.
- Miyamoto, S.; Del Re, D.P.; Xiang, S.Y.; Zhao, X.; Florholmen, G.; Brown, J.H. Revisited and revised: Is RhoA always a villain in cardiac pathophysiology? J. Cardiovasc. Transl. Res. 2010, 3, 330–343.
- Pulinilkunnil, T.; An, D.; Ghosh, S.; Qi, D.; Kewalramani, G.; Yuen, G.; Virk, N.; Abrahani, A.; Rodrigues, B. Lysophosphatidic acid-mediated augmentation of cardiomyocyte lipoprotein lipase involves actin cytoskeleton reorganization. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2802–H2810.
- Siehler, S. Regulation of RhoGEF proteins by G12/13-coupled receptors. Br. J. Pharmacol. 2009, 158, 41–49.
- Ma, X.; Zhao, Y.; Daaka, Y.; Nie, Z. Acute activation of beta2-adrenergic receptor regulates focal adhesions through betaArrestin2- and p115RhoGEF protein-mediated activation of RhoA. J. Biol. Chem. 2012, 287, 18925–18936.
- Lecuona, E.; Ridge, K.; Pesce, L.; Batlle, D.; Sznajder, J.I. The GTP-binding protein RhoA mediates Na,K-ATPase exocytosis in alveolar epithelial cells. Mol. Biol. Cell 2003, 14, 3888–3897.
- Triposkiadis, F.; Karayannis, G.; Giamouzis, G.; Skoularigis, J.; Louridas, G.; Butler, J. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J. Am. Coll. Cardiol. 2009, 54, 1747–1762.
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac beta-Adrenergic Signaling During Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904.
- Cohn, J.N.; Levine, T.B.; Olivari, M.T.; Garberg, V.; Lura, D.; Francis, G.S.; Simon, A.B.; Rector, T. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N. Engl. J. Med. 1984, 311, 819–823.
- Katsuumi, G.; Shimizu, I.; Yoshida, Y.; Hayashi, Y.; Ikegami, R.; Suda, M.; Wakasugi, T.; Nakao, M.; Minamino, T. Catecholamine-Induced Senescence of Endothelial Cells and Bone Marrow Cells Promotes Cardiac Dysfunction in Mice. Int. Heart J. 2018, 59, 837–844.
- Bonnefont-Rousselot, D.; Mahmoudi, A.; Mougenot, N.; Varoquaux, O.; Le Nahour, G.; Fouret, P.; Lechat, P. Catecholamine effects on cardiac remodelling, oxidative stress and fibrosis in experimental heart failure. Redox Rep. 2002, 7, 145–151.
- Myagmar, B.E.; Flynn, J.M.; Cowley, P.M.; Swigart, P.M.; Montgomery, M.D.; Thai, K.; Nair, D.; Gupta, R.; Deng, D.X.; Hosoda, C.; et al. Adrenergic Receptors in Individual Ventricular Myocytes: The Beta-1 and Alpha-1B Are in All Cells, the Alpha-1A Is in a Subpopulation, and the Beta-2 and Beta-3 Are Mostly Absent. Circ. Res. 2017, 120, 1103–1115.

