One of the key proteins involved in stress-mediated cardiomyocyte signal transduction is a small GTPase RhoA. Importantly, the regulation of RhoA activation is critical for effective immune cell response and is being considered as one of the potential therapeutic targets in many immune-cell-mediated inflammatory diseases.
1. Introduction
The immune system is based on two main complexes: (1) the innate immune system and (2) the adaptive immune system. The innate immune system is triggered by direct contact with pathogens or inflammatory/danger signals and includes non-cellular responses, i.e., the release of inflammatory cytokines, and cellular responses, i.e., infiltration of innate immune cells (macrophages, dendritic cells, and granulocytes) into affected tissue [12]. The adaptive immune system also involves non-cellular (hormonal) and cellular responses (stimulation of B- and T-cells), but in contrast to the innate response, it builds up an “immunologic memory” by developing pathogen-specific receptors [12]. RhoA (ras homolog family member A), an ubiquitously expressed small GTPase, acts as a molecular switch not only in the activation of cytoskeletal proteins but also in responding chemokines, cytokines, and growth factors released from both innate and adaptive immune cells [13]. Furthermore, RhoA activation and RhoA-dependent signaling pathways in cardiomyocytes [14] and in immune cells [13,15] have been shown to mediate immune responses, which play an important role in pathogenesis and progression of cardiac dysfunction [12].
2. Links between Cardiac Hypertrophy, Heart Failure, and Immune Cell Activation
In the last decades, the role of the innate and adaptive immune response is being linked with a number of signaling molecules and pathways, including RhoA activation in cardiomyocytes [13,16]. Furthermore, it has been illustrated that different cardiac diseases (e.g., ischemic, hypertensive, and genetic cardiomyopathies) converge in inducing a common immune response that contributes to disease progression [12].
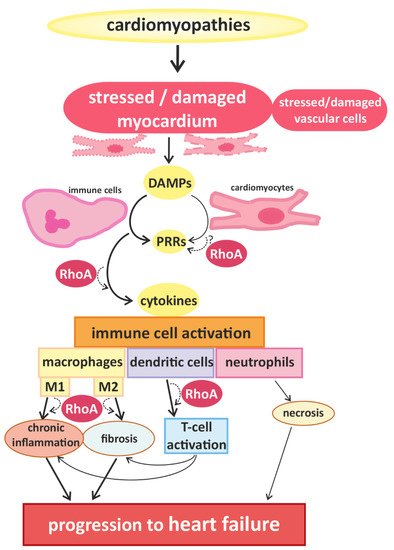
Signals that trigger activation of innate immune cells and subsequent immune responses are pathogen-associated molecules or “danger-molecules” and other signals arising from damaged tissue [12]. The main group of receptors for inflammatory signals consists of pattern recognition receptors (PRRs) [12]. PRRs, which are generally upregulated in HF [17], are commonly expressed by immune cells, but have also been found in cardiac cells [18,19]. Although PPRs are best known for their activation after contact with pathogens, it can also be activated by danger-associated molecular patterns (DAMPs) that are released by damaged or dying cells [20], e.g., injured myocardium. The activation of PPRs triggers the release of inflammatory cytokines [21] (e.g., interferones (IFN) and IL-1β) and thus accelerates immune responses [12] (Figure 1).
Figure 1. Cardiomyopathies of different causality converge in myocardial damage, inducing immune cell responses and promoting disease progression. Cardiomyocytes and blood vessel cells that are stressed or injured release danger signals with “danger-associated molecular patterns” (DAMPs), which are recognized by “pattern recognition receptors” (PRRs), expressed by immune cells and cardiomyocytes. The activation of PRRs induces the migration and activation of (more) immune cells via cytokine signaling. In damaged myocardium, classically activated M1-type and alternatively activated M2-type macrophages are found, which are associated with chronic inflammation and fibrosis, respectively. Furthermore, dendritic cells and co-stimulated T-cells contribute to the development of inflammatory and fibrotic remodeling. Infiltrating neutrophils are associated with increased necrosis. These processes accelerate the progression of cardiac dysfunction. RhoA-dependent signaling is essential for effective immune cell activation and might also play a role in the transduction of danger signals and receptor activation in cardiomyocytes. In addition RhoA signaling is involved in macrophage polarization and signaling pathways mediating the interaction between dendritic cells and T-cells, contributing to pro- and anti-inflammatory remodeling and finally, heart failure.
Natriuretic peptides are key players of cardiac hypertrophy and HF [
14]. Brain and atrial natriuretic peptides (BNP and ANP) are part of the “hypertrophic gene program” in cardiomyocytes and their upregulation, which is partly mediated by RhoA activation, is an established marker for cardiac hypertrophy [
14,
16,
22].
In addition, different forms of programmed cell death have been associated with the development and progression of cardiac diseases [
21,
22]. Besides apoptosis, this includes “programmed necrosis” or necroptosis of myocardial cells [
21,
22,
23].
The best studied immune cells in the context of cardiomyopathies are macrophages. In general, two classes of macrophages are defined: (1) classically activated macrophages (=M1), which correlated with pro-inflammatory processes and (2) alternatively activated macrophages (=M2) correlating with anti-inflammatory processes. Most macrophages found in damaged (cardiac) tissue originate from myeloid naïve monocytes, which circulate in the blood stream, migrate into affected tissue, and differentiate after activation, which belong to the M1 class [
36]. In addition, one subclass of macrophages, called resident macrophages (rcMACs), resides in specific tissues including myocardium under physiological circumstances [
36,
37]. These residential cardiac macrophages can get activated directly by signals of surrounding (cardiac) cells and they belong to the M2 class [
36,
37]. In this context RhoA signaling has been linked to macrophage polarization via its main effector, ROCK1, in mice cells [
38]. Furthermore, activation of macrophages has been demonstrated to correlate with the progression of cardiac hypertrophy and fibrosis to HF [
12,
39,
40,
41] (
Figure 1).
It has been suggested that pro-inflammatory signals in hypertrophic hearts generated by injured myocardium induce differentiation of circulating monocytes to pro-inflammatory M1-type macrophages, and even induce pro-inflammatory responses in residential macrophages, enhancing disease progression [
39,
40]. In addition, an increase in M2-type macrophage polarization in the heart has been linked to progression of hypertrophy to HF, because the anti-inflammatory responses are known to induce fibrosis [
12].
Besides macrophages, dendritic cells (DCs) and neutrophils are the main actors of the innate immune response system. DCs are derived from myeloid progenitors and are located in tissues with contact to the external environment and in the blood. Upon activation, they migrate to lymph nodes, generate and present specific antigen complexes and so activate B- and T-cells. DCs have been found in cardiac tissue injured by cardiac infarction and there is some evidence that they are involved in the subsequent cardiac remodeling [
45,
46]. In this context, a beneficial effect of DC activation in the heart by regulation of monocyte/macrophage homeostasis has been suggested [
46]. In contrast, DCs might also promote the development and progression of cardiac hypertrophy and HF [
47,
48].
Data on links between cells of the adaptive immune system (T- and B-cells) and cardiac diseases, especially hypertrophy, is also insufficient. As mentioned above, there is some evidence that recruitment of T-cells enhances the progress of TAC-induced cardiac hypertrophy [
47,
48]. Furthermore, mice with depletion of T-cells exhibit lower number of infiltrating macrophages, reduced fibrosis, and reduced cardiac dysfunction after TAC [
47]. Nevertheless, more studies are needed to identify interconnections of hypertrophic cardiomyocytes and activation of T- and/or B-cells. Taken together, there is some evidence for links between cardiac hypertrophy and immune system responses. An overlap of risk factors for cardiomyopathies and (chronic) inflammation as well as the activation of immune cells in a variety of cardiac diseases are promising fields for further research, as is the postulated interplay of upregulation of hypertrophic markers (BNP and ANP) and cytokine signaling [
23,
24,
25,
26,
27].
3. RhoA Activation and Signaling in Immune Cells
Recent studies demonstrate that RhoA plays an important role in immune responses, with its effects being highly dependent on the spatio-temporal regulation of RhoA activation in different innate and adaptive immune cells [13,51]. On molecular level, the activation of RhoA in immune cells and cardiomyocytes depends on its change from a guanosine diphosphate (GDP)-bound “inactive” to a guanosine triphosphate (GTP)-bound “active” state and back [52]. This cycle is regulated by regulatory proteins, namely guanine nucleotide exchange factors (GEFs), GTPase activating proteins (GAPs), guanine nucleotide dissociation inhibitors (GDIs) and GDI dissociation factors (GDFs) [16]. GEFs facilitate the dissociation of GDP from RhoA, thus accelerating the binding of GTP and allowing the activation of downstream effectors [53]. GAPs catalyze the hydrolysis of GTP back to GDP and thus the release of the effector [44]. GDIs disconnect RhoA from the plasma membrane, thus inhibiting the dissociation of GDP [54], while GDFs initiate the dissociation of GDIs from RhoA allowing the cycle to start again [16]. A number of these regulators have been shown to be identical in immune cells and cardiomyocytes (e.g., LARG (leukemia-associated RhoGEF), GEF-H1/Lfc (Lbc’s first cousin), PDZ-RhoGEF, p190RhoGAP, and Vav1) [49,55], while others were found specifically in immune cells (e.g., RhoA-GAP, Myo9B, and Rho-GEF7) [56]. Even some of the up- and downstream signaling pathways of RhoA described in cardiomyocytes have been proven to play an important role in immune cells, e.g., RhoA activation via Gα12/13-coupled membrane receptors [57,58,59] and RhoGEF or activation of the RhoA effector, ROCK [13,15,57,58,59,60].
RhoA activation has also been associated with activation of β2-adrenergic receptors (β2-AR), probably via p115RhoGEF [61,62]. Hyperactivity of the sympathetic nervous system is one of the hallmarks of heart failure that involves catecholamine spillover, and is associated with pro-inflammatory signaling [63,64,65]. Several studies have shown that infusion of isoprenaline—a synthetic catecholamine and β2-AR agonist—in mice induces cardiac inflammation and dysfunction [66], and infusion of noradrenalin, also a β2-AR agonist, induces cardiac hypertrophy and fibrosis in rats [67]. However, β1-ARs have been shown to be ubiquitously expressed in rodent (ventricular) cardiomyocytes, while β2-ARs were only found in a very small percentage of these myocytes [68]. In contrast, β2-ARs were found to be abundant in non-myocytes of rodent heart tissue [68], suggesting that endothelial cells, fibroblasts and/or immune cells in cardiac tissue likely express β2-ARs.
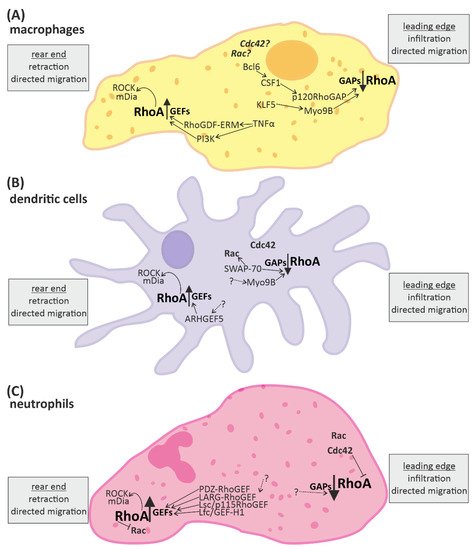
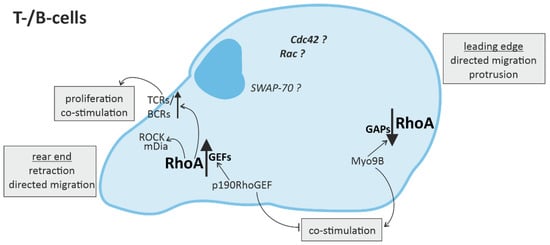
Overall, the processes affected by RhoA in the context of immune responses are fundamental cellular processes such as cytoskeletal arrangement, mobility, cell-cell contact, cell-cycle regulation, proliferation and cell survival, as well as directed migration and (co-) stimulation of immune cells (Figure 2 and Figure 3).
Figure 2. RhoA signaling plays an important role in cells of the innate immune system. Directed migration of macrophages, dendritic cells, and neutrophils is mediated by spatio-temporal regulated RhoA activation. In general, RhoA is activated in the rear end and reduced in the leading edge. The up-regulation of RhoA is mediated by “guanine nucleotide exchange factors” (GEFs) and leads to activation of the downstream effectors ROCK (rho-associated, coiled-coil-containing protein kinase) and mDia (diaphanous-related formin-1), which promote retraction of the rear end. RhoA-downregulation is mediated by “GTPase activating proteins” (GAPs) and facilitates infiltration or protrusion. Some signaling molecules up- and downstream of RhoA activation have been identified in different immune cells. However, many aspects of the signaling pathways are not fully known yet (indicated by dotted arrows and question marks). (A) In macrophages some regulators of RhoA have been identified. KLF5 (Krüppel-like factor-5) and CSF1 (macrophage colony stimulating receptor 1) activation via Bcl6 (B-cell lymphoma-6) lead to activation of the RhoA-GAPs Myo9B and p120RhoGAP, respectively, reducing RhoA activation at the front. Contrarily, the RhoGDF ERM and PI3K (phosphoinositid-3-kinase) promote the activation of RhoA at the end. (B) In dendritic cells the RhoA-GAP Myo9B is also located at the leading edge, attenuating RhoA activation, while the Rho-GEF5 is found in the rear end, promoting RhoA activation. Furthermore, SWAP-70 (switch-associated protein 70) was identified as a regulator of small GTPase activity. (C) In neutrophils a number of GEFs facilitating RhoA activation have been identified. In addition the small GTPases Rac and Cdc42 are located close to the leading edge.
Figure 3. RhoA signaling in cells of the adaptive immune system. Directed migration of T- and B-cells is regulated by spatio-temporal activation of RhoA, with mainly RhoA activation in the rear end and RhoA downregulation in the leading edge. In T- and B-cells, RhoA activation in the rear end not only leads to retraction via ROCK (rho-associated, coiled-coil-containing protein kinase) and mDia (diaphanous-related formin-1), but is also associated with the promotion of T-cell receptor (TCR) or B-cell receptor (BCR) activity, respectively, inducing T-cell proliferation and co-stimulation of more immune cells.
4. Conclusions
Taken together, the existing data on the interplay of cardiomyocytes and immune cells demonstrates good evidence for links between cardiomyocyte damage, immune cell activation and progression of cardiac dysfunction. The intercellular signaling involved in and affected by the development and progression of cardiac diseases includes cardiomyocytes, cells of the innate and adaptive immune system, and other cells present in the myocardium (e.g., fibroblasts). The summary of the data has pointed out that in the intracellular signaling, RhoA is one of the key mediators. Regulation of RhoA activation and RhoA-dependent downstream signaling plays an important role in cardiomyocytes and immune cells. The data on effects of RhoA activation and inhibition is often contradictory, depending on specific cell types, spatial, and temporal regulation. For further research, induction of immune cell responses should be considered when analyzing the mechanisms of the development and progression of cardiomyocyte damage and cardiac dysfunction.
This entry is adapted from the peer-reviewed paper 10.3390/cells10071681