Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Patrizia HRELIA | + 2368 word(s) | 2368 | 2021-07-06 04:10:04 | | | |
| 2 | Peter Tang | Meta information modification | 2368 | 2021-07-19 03:58:27 | | | | |
| 3 | Lindsay Dong | Meta information modification | 2368 | 2021-10-09 11:42:31 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Hrelia, P. NLRP3 and Infections. Encyclopedia. Available online: https://encyclopedia.pub/entry/12129 (accessed on 09 August 2026).
Hrelia P. NLRP3 and Infections. Encyclopedia. Available at: https://encyclopedia.pub/entry/12129. Accessed August 09, 2026.
Hrelia, Patrizia. "NLRP3 and Infections" Encyclopedia, https://encyclopedia.pub/entry/12129 (accessed August 09, 2026).
Hrelia, P. (2021, July 16). NLRP3 and Infections. In Encyclopedia. https://encyclopedia.pub/entry/12129
Hrelia, Patrizia. "NLRP3 and Infections." Encyclopedia. Web. 16 July, 2021.
Copy Citation
Amyloid beta (Aβ)-induced abnormal neuroinflammation is recognized as a major pathological feature of Alzheimer’s disease (AD), which results in memory impairment. Research exploring low-grade systemic inflammation and its impact on the development and progression of neurodegenerative disease has increased. A particular research focus has been whether systemic inflammation arises only as a secondary effect of disease, or it is also a cause of pathology. The inflammasomes, and more specifically the NLRP3 inflammasome, are crucial components of the innate immune system and are usually activated in response to infection or tissue damage.
NLRP3
neuroinflammation
infections
neurodegeneration
Alzheimer’s disease
COVID-19
1. Alzheimer’s Disease and Inflammation
Alzheimer’s disease (AD) is the most common neurodegenerative disease in older people representing the most frequent form of dementia worldwide [1]. Although by 2050, the number of diagnoses is expected to reach 131.5 million, to date, almost all clinical trials have failed—underling the need for further research of novel therapeutic approaches [2].
AD is mainly characterized by two pathological changes, the deposition of extracellular senile plaques of β-amyloid (Aβ) protein, and the formation of intracellular neurofibrillary tangles (NFTs), due to the hyperphosphorylation of tau protein [1]. In physiological conditions, Aβ peptides are removed from the brain tissue both by degradation and by removal into cerebrospinal fluid and blood vessels [3].
The abnormal accumulation of Aβ in the brain, especially the oligomeric form, is an early feature of AD and is usually associated with neuronal loss, inflammatory responses, and oxidative stress [4]. Recently, increasing evidence suggests a central role of the immune system in the progression or even the origin of AD [5]. Many studies highlighted the role of neuroinflammation in the progression of AD, in which the release of inflammatory mediators can influence neuronal cells and their function [6]. Indeed, the inflammatory response has a crucial role in different neurodegenerative diseases, and it is primarily driven by the microglia, which release cytokines causing chronic neuroinflammation [6][7]. Microglia originate from primitive macrophages and are the resident myeloid cells of the central nervous system (CNS). These cells not only check for pathogens and cell debris in the parenchyma of the CNS, but also support homeostasis and brain plasticity, being involved in the formation of neuronal connections during development [8][9][10]. In physiological conditions, microglia play a critical role in several developmental events, as the formation of neural circuits, synaptic pruning, and remodeling, neurogenesis, clearing cellular debris, proteins aggregate, and pathogens [3][11][12][13]. Microglia are the non-neuronal CNS cells most closely related to changes observed in AD. Microglia remove by phagocytosis Aβ oligomers and protofibrils [14]. Aβ oligomers propagate into the brain parenchyma, arousing a stronger microglial response and memory impairment than fibrils [15]. Microglia immune response against Aβ oligomers in the hippocampus may be implicated in the pathogenesis of late-onset AD [16]. However, impairment in this capacity may lead to an adverse increase of Aβ species in the CNS, which could then aggregate further into Aβ plaques. Moreover, Aβ peptide can bind to microglia’s receptors driving the production of proinflammatory cytokines and chemokines [17][18]. Different studies have shown in AD mouse models that the increase of CD68, a marker for microglial activation, is associated with Aβ plaques [19][20]. Furthermore, the fibrillar form of Aβ can induce inflammasome activation in microglia, but less is known about the capacity of small Aβ oligomers and protofibrils, that seem to be more neurotoxic, to activate the inflammasome in microglia [21][22]. The extended exposure of microglia to Aβ can impair their function, decreasing phagocytosis and reducing the capacity to extend processes towards the lesioned tissue with the result that Aβ is inefficiently cleared from the neuronal tissue [23][24]. Different proinflammatory cytokines, like the tumor necrosis factor–α (TNF–α) or interleukin (IL)–1β, –6, –12 and –23, maintain a state of microglial activation and they could trigger each other, leading to a positive feedback loop which accelerates AD pathology [25][26][27]. In particular, IL-1β induces IL-6, which is dramatically increased in AD patients [28]. This last cytokine may have a crucial role in AD, indeed not only a mutation in the gene encoding for IL-6 may result in a late––onset disease, but it can also play a role in the synthesis and expression of amyloid precursor protein (APP) [29]. Moreover, higher levels of IL-1β may affect tau hyperphosphorylation, and thus, aggravate AD pathology, impairing long term potentiation (LTP) and memory formation [30]. In physiological conditions, microglia have a critical role in maintaining a healthy brain. Some structural variants of genes expressed on microglia and encoding for immune receptors, such as TREM2, CD33, and CR1, have been associated with a higher risk of AD [31][32][33]. Moreover, altered gene expression in the regulation of the immune system in AD and its contribution to the pathology, support a pathogenic role of CNS–resident myeloid cells, like microglia, in the evolution of the disease [34][35]. Microglia dysfunction may occur not only by mutations, but also consequently to a long–lasting Aβ exposure [36].
Regardless of the cause, altered microglia lose their beneficial and physiological functions to develop a detrimental, senescent–like phenotype. In addition, when microglia become overactivated or reactive, they can induce detrimental neurotoxic effects releasing numerous cytotoxic elements [37].
Inflammasomes are defined as ‘canonical’ when their assembly requires caspase-1, and as ‘non-canonical’ when their assembly depends on human caspase-4 or caspase-5. As shown in Figure 1, proteins in the NLR family are constituted of a central NOD domain, C–terminal leucine–rich repeats (LRRs), and N–terminal caspase recruitment domains (CARD) or pyrin domains (PYD). These sensors initiate the assembly of canonical inflammasomes by recruiting caspase-1, with or without the ASC adaptor in an ATP–dependent manner [38].
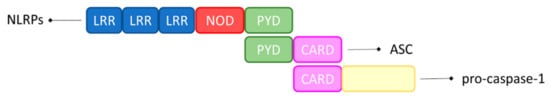
Figure 1. Schematic representation of the inflammasome’s components. Inflammasome contains a C–terminal LRR domain, an N–terminal CARD or PYD domain, and a central NOD domain. ASC consists of an N–terminal PYD and a C–terminal CARD. Once activated, inflammasome acts as a sensor molecule and connects to ASC via the PYD–PYD interaction. Finally, ASC recruits pro-caspase-1 via CARD–CARD interaction, which promotes the self–cleavage and the activation of pro-caspase-1.
2. NLRP3 and Aβ
NLRP3 is ubiquitously expressed in CNS, and it has been found to be highly expressed in the brain of AD patients [39]. To date, no data is available about the possible pharmacological approach in AD based on the inhibition of NLRP3. Misfolded protein aggregates, like Aβ depositions, can promote NLRP3 activation by increasing the expression of the major histocompatibility complex II (MHC–II) on the cell surface [21].
After its activation, the NLRP3 inflammasome increases the release of active caspase-1, and the subsequent secretion of IL-1β and IL-18, which may result in chronic inflammatory responses, neuronal death, and pyroptosis in the CNS [40]. On the other hand, the inhibition of IL-1β signaling may contribute to disease–modifying effects, as shown by the expression of IL-1β in Aβ–plaque, associated with microglial cells [41][42]. Evidence suggests that the levels of active caspase-1 and IL-1β increase in the microglia of AD animal models and patients, and it can be associated with the onset and progression of the pathology [21][27][43]. Moreover, patients with amyloidosis showing cognitive impairments present higher levels of proinflammatory cytokines and lower levels of IL-10 in the serum, as compared to patients without brain amyloidosis [44]. In this view, the activation of microglia in the hippocampus may influence the cytokine profiles in the serum, and this activation may result in a decrease of IL-10 levels [45]. Indeed, emerging evidence indicates that IL-10 may act in a negative feedback loop to regulate the NLRP3 inflammasome during chronic stimulations [46].
An interesting study by Lučiūnaité et al. shows that soluble, low molecular weight Aβ oligomers and protofibrils, with a maximum size of 5 nm, can activate the NLRP3 inflammasome in microglia [47]. The study shows that small Aβ fragments activate murine microglia without altering their viability, suggesting that these species can induce an innate immune response prior to their deposition in amyloid plaques. Moreover, the authors investigate whether soluble Aβ species were able to activate the NLRP3 inflammasome. Indeed, as fibrillary Aβ can act as DAMP and activate the NLRP3 inflammasome, also small soluble Aβ oligomers and protofibrils may activate all components of the NLRP3 inflammasome too, inducing an early neuroinflammatory response [47][48]. Additionally, the activation of the NLRP3 inflammasome boosts Aβ aggregation by reducing phagocytosis [43].
In AD, the presence of Aβ plaques recruits microglia to phagocyte the aggregated forms, especially oligomers and fibrils. This condition induces the activation of the NLRP3 inflammasome, with a subsequent release of proinflammatory cytokines, as IL-1β, and potentially neurotoxic factors. In turn, cytokines and factors released to enhance the neurotoxic effects of Aβ and worsen the pathological processes of AD [49]. It has been described that the NLRP3 inflammasome might be essential for the immune responses in AD. Indeed, Halle et al. showed that Aβ increased the activation of the NLRP3 inflammasome in microglial cells [21]. This hypothesis is also confirmed by Heneka et al., who showed in APP/PS1 mice, that Aβ can activate NLRP3 inflammasome in microglia, inducing an inflammatory M1 phenotype, characterized by an elevated expression of proinflammatory factors, resulting in increased hippocampal and cortical Aβ deposition, neuronal loss, and cognitive impairment. Interestingly, NLRP3 activation in APP/PS1 mice occurs only in microglia associated with the presence of Aβ plaques depositions; underlying that microglia–specific NLRP3 activation contributes to AD pathogenesis. However, in this transgenic model, mice with deletions for NLRP3 or caspase-1 show a reduced impairment in spatial memory abilities and a lower inflammatory response. Moreover, the deletion induces microglia anti-inflammatory M2 phenotype, with decreased caspase-1 and IL-1β secretion, reduced amyloid depositions, and improved cognitive functions [43][50].
Another study conducted by Venegas et al. on APP/PS1 mice showed that the intrahippocampal injection of ASC fragments promotes Aβ plaque formation and accumulation, but it failed to induce Aβ pathology in ASC–deficient mice [51]. Moreover, ASC or NLRP3 deficiencies have been associated with a decreased tau pathology and protected tau transgenic mice against cognitive impairment [52]. Another study has shown that the suppression of IL-1β in the triple transgenic (3 × Tg) mouse model of AD restores cognitive abilities, reduces tau pathology, and reestablishes the function of the neuronal beta–catenin pathway [41]. In this view, the specific abnormal activation of NLRP3 in microglia induces chronic neuroinflammation in AD, leading to microglial Aβ phagocytic dysfunction and neuronal damage. However, this process might be altered by a damage to the inflammasome. In summary, clarifying the links between innate immune activation and microglia–dependent NLRP3 inflammasome activation may explain the functional role of NLRP3 in AD. Its regulation may reduce neuroinflammation in AD, and therefore, be a novel therapeutic strategy for this disease.
A recent study demonstrates that NLRP3 inflammasome plays a critical role in a mouse model of sporadic AD. Results showed that the intracerebroventricular (icv) injection of streptozotocin (STZ) activated the NLRP3 inflammasome, reduced Aβ clearance, and induced neuronal loss and cognitive impairment. Moreover, the inflammatory response enhanced the activation of NLRP3, amplifying the microglial reaction, and worsening the pathological damage. Interestingly, the inhibition or depletion of microglial NLRP3 reversed these effects [53].
All these findings highlighted a fundamental role of the NLRP3 inflammasome in the progression of AD and suggested that the pharmacological inhibition of the NLRP3 may represent a turning point in treating neurodegenerative diseases.
3. NLRP3 and Infections
A multitude of viruses can cause severe diseases, such as hepatitis C virus (HCV), human immunodeficiency virus–1 (HIV–1), and influenza A virus (IAV). For this reason, the host has evolved highly conserved sensors, named PRRs, to remove invading viruses activating antiviral immune response [54]. Moreover, the role of the NLRP3 inflammasome is essential for the antiviral immune responses. Indeed, several viruses induce early activation of NLRP3, which reduces viral replication and decreases mortality in mouse models [55]. In physiological conditions, NLRP3 levels are low to prevent aberrant inflammasome activation. In the case of viral infection, NF–κB signaling is activated through PRRs–dependent pathways, which induce IFN–β or TNF–α activation that, in turn, activate NF–κB to initiate the NLRP3 inflammasome response [56]. To the best of our knowledge, a specific ligand able to bind directly to NLRP3 is not known. The activation of the inflammasome is usually associated with PAMPs and DAMPs. Interestingly, also small viral components could activate the NLRP3 inflammasome inducing IL-1β secretion in macrophages [57]. ROS formation and cellular homeostasis are fundamental for the NLRP3 inflammasome activation, as potassium (K+) or calcium (Ca2+) efflux or influx are established activators that lead to mitochondria damage and ROS formation, potentiating the NLRP3 inflammasome activation [58][59][60].
We have already explained as Aβ formation may induce the NLRP3 inflammasome activation. Interestingly, the open reading frame 8b (ORF8b) of the severe acute respiratory syndrome coronavirus–2 (SARS–CoV–2) forms intracellular aggregates that represent a danger signal able to induce endoplasmic reticulum stress and lysosomal damage, resulting in the NLRP3 inflammasome activation [61]. Several studies showed that the NLRP3 inflammasome and IL-1β are implicated in the inflammatory response during lung injury and acute respiratory distress syndrome (ARDS) [62][63]. Indeed, middle east respiratory syndrome–related coronavirus (MERS CoV), SARS–CoV, and influenza patients with ARDS not only show higher levels of IL-1β in bronchoalveolar fluid and plasma as compared to healthy controls, but this condition is also associated with worse clinical outcomes [64][65][66]. Indeed, the aberrant activation of NLRP3 and downstream mediators often lead to pathological tissue injury during infection [67]. For example, several studies have highlighted its important role in relation to the pathogenesis of ARDS, which is driven by the same proinflammatory cytokines released by the inflammasome [68]. Interestingly, animals lacking in inflammasome’s components showed reduced lung injury and increased survival rate following influenza infection [69]. A recent study by Blanco–Melo et al. demonstrated that SARS–CoV–2 infection induced the expression of many cytokines and chemokines, including TNF–α, IL-6, and IL-1β, contributing to the tissue damage [70]. Even more interestingly, this pathological immune response is characterized by a hyperinflammatory microenvironment limited to the site of tissue injury. With the development of the inflammatory cascade, IL-1β and TNF–α induce the secretion of further NLRP3 cytokines, such as IL-6, which, owing to the loss of vascular integrity, can be detected in the peripheral blood and may activate the NLRP3 inflammasome in other immunological pathways [71][72].
In conclusion, NLRP3 activation and associated inflammation are a double–edged sword in the antiviral host defense. The modulation of NLRP3 inflammasome activity may be a promising approach to counteract viral diseases and the subsequent inflammatory reactions.
References
- Alzheimer’s Association. 2021 Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2021, 17, 327–406.
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75.e2.
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236.
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 inflammasome activation: Its role in the treatment of Alzheimer’s disease. Neurochem. Res. 2020, 45, 2560–2572.
- Heneka, M.; Kummer, M.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477.
- Ardura-Fabregat, A.; Boddeke, E.W.G.M.; Boza-Serrano, A.; Brioschi, S.; Castro-Gomez, S.; Ceyzériat, K.; Dansokho, C.; Dierkes, T.; Gelders, G.; Heneka, M.T.; et al. Targeting neuroinflammation to treat Alzheimer’s disease. CNS Drugs 2017, 31, 1057–1082.
- Giannoni, P.; Arango-Lievano, M.; Neves, I.D.; Rousset, M.C.; Baranger, K.; Rivera, S.; Jeanneteau, F.; Claeysen, S.; Marchi, N. Cerebrovascular pathology during the progression of experimental Alzheimer’s disease. Neurobiol. Dis. 2016, 88, 107–117.
- Sarlus, H.; Heneka, M.T. Microglia in Alzheimer’s disease. J. Clin. Invest. 2017, 127, 3240–3249.
- Hammond, T.R.; Robinton, D.; Stevens, B. Microglia and the brain: Complementary partners in development and disease. Annu. Rev. Cell Dev. Biol. 2018, 34, 523–544.
- Chen, Z.; Trapp, B.D. Microglia and neuroprotection. J. Neurochem. 2016, 136, 10–17.
- Diaz-Aparicio, I.; Paris, I.; Sierra-Torre, V.; Plaza-Zabala, A.; Rodríguez-Iglesias, N.; Márquez-Ropero, M.; Beccari, S.; Huguet, P.; Abiega, O.; Alberdi, E.; et al. Microglia actively remodel adult hippocampal neurogenesis through the phagocytosis secretome. J. Neurosci. 2020, 40, 1453–1482.
- Ortega-Martinez, S.; Palla, N.; Zhang, X.; Lipman, E.; Sisodia, S.S. Deficits in enrichment-dependent neurogenesis and enhanced anxiety behaviors mediated by expression of Alzheimer’s disease-linked Ps1 variants are rescued by microglial depletion. J. Neurosci. 2019, 39, 6766–6780.
- Weinhard, L.; Di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228.
- Lee, C.Y.D.; Landreth, G.E. The role of microglia in amyloid clearance from the AD brain. J. Neural Transm. 2010, 117, 949–960.
- He, Y.; Zheng, M.M.; Ma, Y.; Han, X.J.; Ma, X.Q.; Qu, C.Q.; Du, Y.F. Soluble oligomers and fibrillar species of amyloid β-peptide differentially affect cognitive functions and hippocampal inflammatory response. Biochem. Biophys. Res. Commun. 2012, 429, 125–130.
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405.
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 mediates the innate host response to β-amyloid. J. Exp. Med. 2003, 197, 1657–1666.
- Patel, N.S.; Paris, D.; Mathura, V.; Quadros, A.N.; Crawford, F.C.; Mullan, M.J. Inflammatory cytokine levels correlate with amyloid load in transgenic mouse models of Alzheimer’s disease. J. Neuroinflammation 2005, 2, 9.
- Damjanac, M.; Bilan, A.R.; Barrier, L.; Pontcharraud, R.; Anne, C.; Hugon, J.; Page, G. Fluoro-Jade® B staining as useful tool to identify activated microglia and astrocytes in a mouse transgenic model of Alzheimer’s disease. Brain Res. 2007, 1128, 40–49.
- Verbeek, M.M.; Otte-Höller, I.; Wesseling, P.; Van Nostrand, W.E.; Sorg, C.; de Waal, R.M.W.; Ruiter, D.J. A lysosomal marker for activated microglial cells involved in Alzheimer classic senile plaques. Acta Neuropathol. 1995, 90, 493–503.
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865.
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large soluble oligomers of amyloid β-protein from alzheimer brain are far less neuroactive than the smaller oligomers to which they dissociate. J. Neurosci. 2017, 37, 152–163.
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective β-amyloid clearance pathways in aging alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360.
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS ONE 2013, 8, e60921.
- Fillit, H.; Ding, W.; Buee, L.; Kalman, J.; Altstiel, L.; Lawlor, B.; Wolf-Klein, G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci. Lett. 1991, 129, 318–320.
- Vom Berg, J.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s diseasea-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819.
- Dursun, E.; Gezen-Ak, D.; Hanağasi, H.; Bilgiç, B.; Lohmann, E.; Ertan, S.; Atasoy, I.L.; Alaylioğlu, M.; Araz, Ö.S.; Önal, B.; et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J. Neuroimmunol. 2015, 283, 50–57.
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421.
- Vallières, L.; Rivest, S. Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1β. J. Neurochem. 1997, 69, 1668–1683.
- Griffin, W.S.T.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflammation 2006, 3, 5.
- Guerreiro, R.; Wojtas, A.; Bras, J.; Carrasquillo, M.; Rogaeva, E.; Majounie, E.; Cruchaga, C.; Sassi, C.; Kauwe, J.S.K.; Younkin, S.; et al. TREM2 variants in Alzheimer’s disease. N. Engl. J. Med. 2013, 368, 117–127.
- Bradshaw, E.M.; Chibnik, L.B.; Keenan, B.T.; Ottoboni, L.; Raj, T.; Tang, A.; Rosenkrantz, L.L.; Imboywa, S.; Lee, M.; Von Korff, A.; et al. CD33 Alzheimer’s disease locus: Altered monocyte function and amyloid biology. Nat. Neurosci. 2013, 16, 848–850.
- Thambisetty, M.; An, Y.; Nalls, M.; Sojkova, J.; Swaminathan, S.; Zhou, Y.; Singleton, A.B.; Wong, D.F.; Ferrucci, L.; Saykin, A.J.; et al. Effect of complement CR1 on brain amyloid burden during aging and its modification by APOE genotype. Biol. Psychiatry 2013, 73, 422–428.
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372.
- Webers, A.; Heneka, M.T.; Gleeson, P.A. The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol. Cell Biol. 2020, 98, 28–41.
- Baik, S.H.; Kang, S.; Lee, W.; Choi, H.; Chung, S.; Kim, J.I.; Mook-Jung, I. A breakdown in metabolic reprogramming causes microglia dysfunction in Alzheimer’s disease. Cell Metab. 2019, 30, 493–507.
- Block, M.L.; Hong, J.-S. Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Prog. Neurobiol. 2005, 76, 77–98.
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022.
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 inflammasome in neurological diseases, from functions to therapies. Front. Cell. Neurosci. 2017, 11, 63.
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219.
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates Tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549.
- Tan, M.S.; Yu, J.T.; Jiang, T.; Zhu, X.C.; Tan, L. The NLRP3 inflammasome in alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882.
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678.
- Cattaneo, A.; Cattane, N.; Galluzzi, S.; Provasi, S.; Lopizzo, N.; Festari, C.; Ferrari, C.; Guerra, U.P.; Paghera, B.; Muscio, C.; et al. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol. Aging 2017, 49, 60–68.
- Fekete, C.; Vastagh, C.; Dénes, Á.; Hrabovszky, E.; Nyiri, G.; Kalló, I.; Liposits, Z.; Sárvári, M. Chronic amyloid β-oligomer infusion evokes sustained inflammation and microglial changes in the rat hippocampus via NLRP3. Neuroscience 2019, 405, 35–46.
- Ip, W.K.E.; Hoshi, N.; Shouval, D.S.; Snapper, S.; Medzhitov, R. Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 2017, 356, 513–519.
- Lučiūnaitė, A.; McManus, R.M.; Jankunec, M.; Rácz, I.; Dansokho, C.; Dalgėdienė, I.; Schwartz, S.; Brosseron, F.; Heneka, M.T. Soluble Aβ oligomers and protofibrils induce NLRP3 inflammasome activation in microglia. J. Neurochem. 2020, 155, 650–661.
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat. Immunol. 2013, 14, 812–820.
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; De Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724.
- Goldmann, T.; Tay, T.L.; Prinz, M. Love and death: Microglia, NLRP3 and the Alzheimer’s brain. Cell Res. 2013, 23, 595–596.
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks crossseed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361.
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673.
- He, X.; Xu, J.; Li, G.; Li, M.; Li, L.; Pei, Z.; Zhang, L.; Hu, X. NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2020, 11, 1–11.
- Tan, X.; Sun, L.; Chen, J.; Chen, Z.J. Detection of microbial infections through innate immune sensing of nucleic acids. Annu. Rev. Microbiol. 2018, 72, 447–478.
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.Y. The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity 2009, 30, 556–565.
- Bauernfeind, F.; Horvath, G.; Stutz, A.; Alnemri, E.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.; Fitzgerald, K.; et al. NF-kB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 2009, 183, 787–791.
- Muruve, D.A.; Pétrilli, V.; Zaiss, A.K.; White, L.R.; Clark, S.A.; Ross, P.J.; Parks, R.J.; Tschopp, J. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 2008, 452, 103–107.
- Pétrilli, V.; Papin, S.; Dostert, C.; Mayor, A.; Martinon, F.; Tschopp, J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007, 14, 1583–1589.
- Nieto-Torres, J.L.; Verdiá-Báguena, C.; Jimenez-Guardeño, J.M.; Regla-Nava, J.A.; Castaño-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339.
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019, 10, 50.
- Shi, C.S.; Nabar, N.R.; Huang, N.N.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019, 5, 101.
- Olman, M.A.; White, K.E.; Ware, L.B.; Simmons, W.L.; Benveniste, E.N.; Zhu, S.; Pugin, J.; Matthay, M.A. Pulmonary edema fluid from patients with early lung injury stimulates fibroblast proliferation through IL-1β-induced IL-6 expression. J. Immunol. 2004, 172, 2668–2677.
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J. Clin. Invest. 2001, 107, 1529–1536.
- Kim, K.S.; Jung, H.; Shin, I.K.; Choi, B.R.; Kim, D.H. Induction of interleukin-1 beta (IL-1β) is a critical component of lung inflammation during influenza A (H1N1) virus infection. J. Med. Virol. 2015, 87, 1104–1112.
- Alosaimi, B.; Hamed, M.E.; Naeem, A.; Alsharef, A.A.; AlQahtani, S.Y.; AlDosari, K.M.; Alamri, A.A.; Al-Eisa, K.; Khojah, T.; Assiri, A.M.; et al. MERS-CoV infection is associated with downregulation of genes encoding Th1 and Th2 cytokines/chemokines and elevated inflammatory innate immune response in the lower respiratory tract. Cytokine 2020, 126, 154895.
- He, L.; Ding, Y.; Zhang, Q.; Che, X.; He, Y.; Shen, H.; Wang, H.; Li, Z.; Zhao, L.; Geng, J.; et al. Expression of elevated levels of pro-inflammatory cytokines in SARS-CoV-infected ACE2+ cells in SARS patients: Relation to the acute lung injury and pathogenesis of SARS. J. Pathol. 2006, 210, 288–297.
- da Costa, L.S.; Outlioua, A.; Anginot, A.; Akarid, K.; Arnoult, D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis. 2019, 10, 346.
- Li, D.; Ren, W.; Jiang, Z.; Zhu, L. Regulation of the NLRP3 inflammasome and macrophage pyroptosis by the p38 MAPK signaling pathway in a mouse model of acute lung injury. Mol. Med. Rep. 2018, 18, 4399–4409.
- Zhang, H.; Luo, J.; Alcorn, J.F.; Chen, K.; Fan, S.; Pilewski, J.; Liu, A.; Chen, W.; Kolls, J.K.; Wang, J. AIM2 inflammasome is critical for influenza-induced lung injury and mortality. J. Immunol. 2017, 198, 4383–4393.
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020, 181, 1036–1045.e9.
- Fung, S.; Yuen, K.; Ye, Z.; Chan, C.; Jin, D. A tug-of-war between severe acute respiratory syndrome coronavirus 2 and host antiviral defence: Lessons from other pathogenic viruses. Emerg. Microbes Infect. 2020, 9, 558–570.
- Fu, Y.; Cheng, Y.; Wu, Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol. Sin. 2020, 35, 266–271.
More
Information
Subjects:
Toxicology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
832
Revisions:
3 times
(View History)
Update Date:
09 Oct 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No